En ytterligare undersökning av de läckta EMA-e-postmeddelandena och konfidentiella Pfizer-BioNTech COVID-19-vaccinrelaterade dokument
I juni publicerade Trial Site News en omfattande utredningsrapport om de läckta e-postmeddelandena från Europeiska läkemedelsmyndigheten (EMA) och andra Pfizer-relaterade konfidentiella rapporter, som avslöjade oroande fakta i samband med godkännandet av Pfizer-BioNTechs covid-19-vaccin. Den avslöjade:
- En politiskt driven kapplöpning mellan viktiga tillsynsmyndigheter i deras brådska att godkänna vaccinet.
- I slutet av november 2020 var tillsynsmyndigheterna, inklusive amerikanska FDA, Europeiska läkemedelsmyndigheten, Health Canada och brittiska MHRA, alla medvetna om den betydande förlusten av RNA-integritet hos de kommersiella partierna (~ 55% mRNA-integritet) av Pfizer-BioNTech-vaccinet jämfört med de kliniska partierna (~ 78% mRNA-integritet). Detta klassificerades av EMA som en ”större invändning” tillsammans med observerade synliga partiklar, som klassificerades som ”orenheter.”
- En läckt PowerPoint-presentation från ett möte mellan Pfizer-BioNTech och EMA den 26 november visade hur denna viktiga invändning på ett chockerande sätt ’löstes’ – specifikationen för RNA-integritet sänktes helt enkelt till 50%, vilket innebar att hälften av alla mRNA-molekyler i de kommersiella satserna tilläts vara trunkerade (inte intakta).
- De potentiella konsekvenserna av förlusten av RNA-integritet när det gäller säkerhet och effektivitet var okända.
Denna rapport fokuserar på ytterligare läckta e-postmeddelanden med särskild hänvisning till EU-kommissionären Ursula von der Leyen och den ovanliga utsträckning som hon var beredd att gå för att få medlemsländerna att undvika att använda artikel 5 (2) (deras nationella Emergency Use Authorisation, tillstånd för användning i nödsituationer, för COVID-19-vacciner) men gå med på ett villkorligt marknadsföringstillstånd (CMA) från EU. Den belyser andra läckta känsliga EMA-dokument: BWP:s (arbetsgruppen för biologiska läkemedel) presentation av kvalitetsbyråns CMC-observationer den 24 november 2020 och Rapporteur’s Rolling Review, som avslöjar mer bevis som stöder de ’större invändningar’ som diskuterades i den ursprungliga Trial Site News-rapporten. Den undersöker oredigerade versioner av Pfizers vaccinkontrakt och EU:s förhandsavtal som undertecknades i november 2020 med Pfizer och BioNTech ’för utveckling, produktion, prioriterade inköpsalternativ och leverans av ett framgångsrikt COVID-19-vaccin för EU:s medlemsstater.’
Känd för skandaler
Ursula von der Leyen har varit föremål för intensiv offentlig granskning på grund av sina privata förhandlingar om ett vaccinavtal värt flera miljarder euro (EU:s största kontrakt) via hemliga sms och telefonsamtal med Pfizers vd Albert Bourla, i strid med kommissionens beslut om en styrgrupp som ska ’ge vägledning under hela utvärderingsprocessen.’ Dessutom vägrade hon att ge allmänheten tillgång till de hemliga textmeddelanden som utväxlats mellan Bourla och henne själv, när hon uppmanades att göra det. Europeiska revisionsrätten offentliggjorde en alarmerande rapport i vilken det sades att ’vi bad kommissionen att förse oss med information om de preliminära förhandlingarna om detta avtal (vetenskapliga experter som konsulterats och råd som erhållits, tidpunkten för samtalen, protokoll från diskussionerna och detaljer om de överenskomna villkoren). Någon sådan information kom dock inte.’
Även Pfizers Bourla fick det hett om öronen när han kallades till Europaparlamentets särskilda utskott för covid-19 för att svara på frågor om de hemliga vaccinavtalen, men drog sig i sista minuten ur mötet med utskottet.
De läckta e-postmeddelandena
Den ursprungliga undersökningsrapporten från Trial Site News belyste det enorma tryck som EC (Europeiska kommissionen) utövade på EMA för att bevilja CMA under en mycket accelererad tidslinje. Ett e-postmeddelande från Noel Wathion (EMA:s tidigare vice verkställande direktör) publicerades, som avslöjade ett ’ganska spänt’ TC (telefonkonferenssamtal) med EU-kommissionären (Ursula von der Leyen) som ’ibland till och med var lite obehagligt.’
Nedan följer ett e-postmeddelande från Hilde Boone på EMA, där hon uppger att ’hon [von der Leyen] kommer att vara beredd att personligen ringa relevanta hälsoministrar för att undvika användningen av artikel 5.2.’
’En försening på flera veckor … var inte lätt att acceptera för EC [Europeiska kommissionen]’, säger Wathion. Han avslöjar också ’hur politiska konsekvenser verkar vara för höga även om den ”tekniska” nivån i medlemsstaterna skulle kunna försvara en sådan ”försening” för att göra resultatet av den vetenskapliga granskningen så robust som möjligt.’

Bildtext:
Kära Olga & Florian
Bara en heads-up: vi har precis avslutat TC mellan kommissionären och ECDC/EMA där kommissionären ställde frågor om det förväntade godkännandet av Pfizer-vaccinet och tidpunkten för FDA vs EU-godkännande. Så automatiskt kom frågan om nationell artikel 5.2 vs CMA upp, vilket Noel förklarade i detalj samt hur vi planerar att ytterligare diskutera det med de nationella behöriga myndigheterna nästa vecka och i HMA. Guido föreslog också att vi skulle överväga att ta upp frågan med hälsoministrarna vid EPSCO.
Sandra och Giorgios sa att de skulle diskutera vidare även inom SANTE, så därav mitt mejl till dig.
(Andrzej var också närvarande)
Också att notera:
Kommissionären sa att eftersom EC gjorde ett åtagande gentemot medlemsstaterna och EP att vaccinerna kommer att finnas tillgängliga för alla medlemsstater samtidigt – och att det därför kommer att vara viktigt att medlemsstaterna inte kommer att ’tvingas’ att använda den nationella vägen på grund av” förseningar” i det formella godkännandeförfarandet. Hon sa också att hon kommer att vara beredd att ringa relevanta hälsoministrar personligen för att undvika användningen av artikel 5.2.
Med vänliga hälsningar,
Hilde
Artikel 5 (2) mot CMA
I artikel 5.2 i direktiv 2001/83 anges att ’Medlemsstaterna får tillfälligt tillåta distribution av ett icke godkänt läkemedel som svar på misstänkt eller bekräftad spridning av patogener, toxiner, kemiska ämnen eller kärnstrålning som alla kan orsaka skada.’ Med andra ord är det motsvarigheten till ett godkännande för användning i nödsituationer på medlemsstatsnivå. Det kan göras mycket snabbt eftersom läkemedlet inte behöver gå igenom den nationella standardprocessen för godkännande.
En expert på EU-lagstiftning förklarade för Trial Site News att nackdelen med att medlemsstaterna använder Art. 5.2, är att det skulle ha gett upphov till konkurrens mellan medlemsstaterna, vilket skulle ha lett till en ojämlik tillgång/distribution av covid-19-vaccinerna, särskilt för de medlemsstater som föredrog att vänta på EU:s CMA (vilket tar längre tid eftersom det ska följa ett kontrollerat och robust ramverk med skyddsåtgärder). Ett CMA från EU bidrog till att säkerställa att vaccinerna skulle ha varit tillgängliga för alla medlemsländer samtidigt, vilket var ett av målen med EU:s covid-19-strategi.
Men kan det ha funnits ytterligare skäl till att von der Leyen var så desperat att få medlemsstaterna att undvika artikel 5.2 att hon var villig att själv ringa alla relevanta hälsoministrar?
I e-postmeddelandet nedan från Noel Wathion säger han ’eftersom det första alternativet fortfarande är att sikta på en CMA snarare än en artikel 5 (2) kopplad eller inte med en artikel 5 (3) ..’

Bildtext:
Kära Thomas,
Jag cc Emer i mitt svar. Som sådant har inte så mycket förändrats eftersom hon tydligt säger ”om allt går bra”. Och ja, detta är i linje med EMA och de uttalanden vi har gjort i flera intervjuer sedan det ögonblick då de preliminära analysresultaten för Pfizer / BioNTech-vaccinet (bekräftat denna vecka av de slutliga analysresultaten) och de preliminära analysresultaten för det andra mRNA-vaccinet för Moderna har offentliggjorts. Det nya enligt min mening är att hon tydligt identifierar de två vacciner som kan komma att godkännas före årets slut. Det finns fortfarande problem med båda (CMC verkar vara ett problem för Pfizer/BioNTech, och den rullande granskningen av Moderna har just inletts, vilket ger mindre tid för granskning) så det återstår att se om allt detta kan lösas i tid, utan att äventyra granskningens robusthet. Vi är medvetna om att de rättsliga verktygen i USA och EU skiljer sig åt, och eftersom alternativ 1 fortfarande är att sträva efter en CMA snarare än en artikel 5.2 kombinerat eller inte med artikel 5.3, försöker vi göra CMA-processen så anpassad som möjligt till pandemisituationen eftersom den hittills främst använts för onkologiläkemedel. Diskussionen vid EU Exe SG i onsdags var mycket användbar och vi påskyndar detta arbete. Detta för mig till nästa punkt, dvs. hur vi bäst säkerställer att anpassningen fortsätter inom nätverket. Vi har nu följande möten inplanerade under de kommande veckorna för att diskutera covid-19:
- HMA på torsdag: 10 minuters uppdatering av MA och ett särskilt ämne om kliniska prövningar, om jag inte misstar mig, som ska presenteras av PEI.
- EU Exe SG veckan efter nästa, där punkten om att göra MA-processen så effektiv som möjligt återigen finns på dagordningen.
- MB den 16-17/12.Som en reflektionspunkt, eftersom vi har att göra med en extraordinär situation av ytterst vikt för nätverket och dess trovärdighet, som ska hanteras under de kommande 6 veckorna (vi kommer att vara i hotspot för de 1a vaccinerna, trycket bör vara mycket mindre säkert med denna nivå av effektivitet och på villkor att det inte finns några större kvalitets- och säkerhetsfrågor), borde vi inte organisera ett brådskande ad hoc-möte mellan HMA, EMA och EC (på cirka 3 timmar) för att ha en mer djupgående diskussion om alla flaskhalsar för att påskynda CMA-processen och även för att anpassa oss till kommunikationen för de olika scenarierna (tidigare godkännande av FDA/ MHRA, liknande tidslinjer, balans mellan snabbhet och robusthet)? Eller alternativt att förlänga HMA-mötet nästa torsdag, eller EU Exe S veckan därpå? Men jag skulle inte vänta mycket längre än till onsdag nästa vecka. Jag ser fram emot din feedback.
KR,
Noel (Slut på bildtext)
I artikel 5.3 anges följande: ’Medlemsstaterna skall fastställa bestämmelser för att säkerställa att innehavare av godkännande för försäljning, tillverkare och hälso- och sjukvårdspersonal inte kan ställas till civilrättsligt eller administrativt ansvar för följder av användningen av ett läkemedel..’
Det faktum att Wathion påpekar att Art. 5 (2) kan kombineras eller inte med Art. 5 (3), väcker den viktiga frågan om medlemsstaterna skulle ha haft möjlighet att inte ge ansvarsimmunitet till innehavarna av godkännandet för försäljning (i detta fall BioNTech) och tillverkarna (BioNTech och Pfizer).
De skadliga Pfizer-kontrakten och förhandsöverenskommelserna om köp
Med tanke på vad vi vet om de läckta Pfizer-kontrakten som gjorts tillgängliga av den ideella konsumentorganisationen Public Citizen, har detta läkemedelsföretag till synes mobbat länder till tystnad, kan ge sig på suveräna staters tillgångar (enligt undantaget från suverän immunitet) och åtnjuta full ’indemnity’ – befrielse från juridiskt ansvar som kan uppstå på grund av deras produkt – i själva verket är det köparen som görs ansvarig å deras vägnar. Detta innebär att regeringar har varit tvungna att betala ersättning till medborgare som har drabbats av en vaccinbiverkan, inte vaccintillverkaren.
Skärmdumpen nedan är hämtad från den oredigerade versionen av förhandsavtalet (APA) mellan Europeiska kommissionen (EC), som agerar på uppdrag av och i medlemsländernas namn, Pfizer Inc. och BioNTech (tillsammans kallade ’The Contractor, entreprenören’) som undertecknades i november 2020 (ungefär samtidigt som de läckta EMA-e-post-meddelandena genererades). I avtalet anges att ’varje deltagande medlemsstat ska säkra och hålla The Contractor och deras dotterbolag skadeslösa från och mot allt ansvar som uppstår i samband med skador, skadestånd och förluster som uppstår från eller i samband med användningen och spridningen av vaccinerna..’

Bildtext:
1.12 SKADESTÅNDSANSVAR
1.12.1 Kommissionen förklarar på de deltagande medlemsstaternas vägnar att användningen av vacciner som framställts inom ramen för detta förhandsavtal kommer att ske under epidemiska förhållanden som kräver sådan användning och att administreringen av vacciner därför kommer att ske på de deltagande medlemsstaternas eget ansvar. Varje deltagande medlemsstat ska därför ge uppdragstagaren, deras närstående företag, underleverantörer, licensgivare och underlicenstagare samt deras tjänstemän, direktörer, anställda och andra ombud och företrädare (tillsammans ”de ersättningsskyldiga personerna”) immunitet från och mot alla uppkomna skulder, uppgörelser enligt artikel I.12.6 och rimliga direkta externa rättsliga kostnader som uppkommit vid försvar av Tredjepartsanspråk (inklusive skäliga advokatarvoden och andra kostnader) avseende skada, skadestånd och förluster enligt definitionen i artikel 1.12.2 (tillsammans ”Förlusterna”) som uppkommit från eller i samband med användning och distribution av Vaccinerna inom den Deltagande medlemsstatens jurisdiktion i fråga. Denna artikel I.12 gäller för förluster som uppkommer från eller hänför sig till vaccinerna (Slut på bildtext)
Information som publicerats på Europeiska kommissionens webbplats anger dock att enligt ett villkorligt EU-godkännande för försäljning (CMA) ’ligger ansvaret hos innehavaren av godkännandet för försäljning’ ( vilket i detta fall är BioNTech). Detta står i direkt konflikt med den APA som EU-kommissionen undertecknade med Pfizer och BioNTech (för 200 miljoner vaccindoser till ett pris av 15,50 euro per dos exklusive moms), en månad innan CMA beviljades. Det är värt att notera att EC publicerade en kraftigt redigerad version av den identiska APA, alla avsnitt som rör skadestånd/ansvar eller som helt enkelt anses vara ’känsliga’ har censurerats.
En annan viktig punkt att ta hänsyn till när det gäller artikel 5.2 vs en EU CMA är att med tanke på att vissa europeiska länder krävde vaccinet för vuxna, högrisksgrupper och vissa arbetssektorer – är det mycket osannolikt att dessa medlemsstater skulle ha kunnat göra det med ett icke godkänt läkemedel, vilket dessa vacciner skulle ha klassificerats enligt artikel 5.2.
CMC:s problem
Wathions e-postmeddelande från den 20 november 2020 tar upp flera oroande punkter – ’det finns fortfarande problem med båda (CMC verkar vara ett problem för Pfizer/BioNTech och den rullande granskningen för Moderna började precis, vilket ger mindre tid till granskning) så det återstår att se om allt detta kan ordnas i tid, utan att äventyra granskningens robusthet.’
I den ursprungliga Trial Site News-rapporten diskuterades vilka CMC-problem (Chemistry Manufacturing and Controls) Pfizer/BioNTech hade, särskilt förlusten av RNA-integritet i de kommersiella batcherna och de okända synliga partiklar som observerats. Wathions talande formulering ’det finns fortfarande problem’ tyder på att dessa ’problem’ inte var lösta utan hade pågått under en längre tid. Wathions oro för att ’äventyra granskningens robusthet’ på grund av behovet att godkänna ’i tid’ betonas. Denna oro för att snabbhet går före säkerhet återspeglas i andra läckta e-postmeddelanden, särskilt från Wathion.
Rapporten innehöll också ett läckt e-postmeddelande från Veronika Jekerle, EMA:s chef för Pharmaceutical Quality Office, där hon beskrev de tre överenskomna större invändningarna och slutsatsen från BWP (Biologics Working Party) om Pfizer-BioNTech-vaccinet. Hennes e-postmeddelande skickades den 24e november, samma dag som BWP-presentationen. Nedan finns en serie skärmdumpar av den faktiska läckta BWP-power point-presentationen (som Jekerle hänvisar till i sitt e-postmeddelande), med titeln ’EMA Quality Office CMC Observations’. I slutet av sitt e-postmeddelande tackar hon ’Ton, Brian och Claudio.’ Ton van der Stappen och Brian Dooley nämns i skärmdumpen av bilden nedan.

I skärmdumpen nedan presenteras omfattande data som backar upp en av EMA:s viktigaste invändningar med hänvisning till den betydande minskningen av %RNA-integriteten mellan de kliniska och kommersiella batcherna. Det är anmärkningsvärt att batcher från Pfizers anläggningar i Andover, USA (~62%) och Puurs, Belgien (~55%) rapporterades ha betydligt lägre %RNA-integritet än batcher som levererades av BNT (BioNTech) och Polymun.
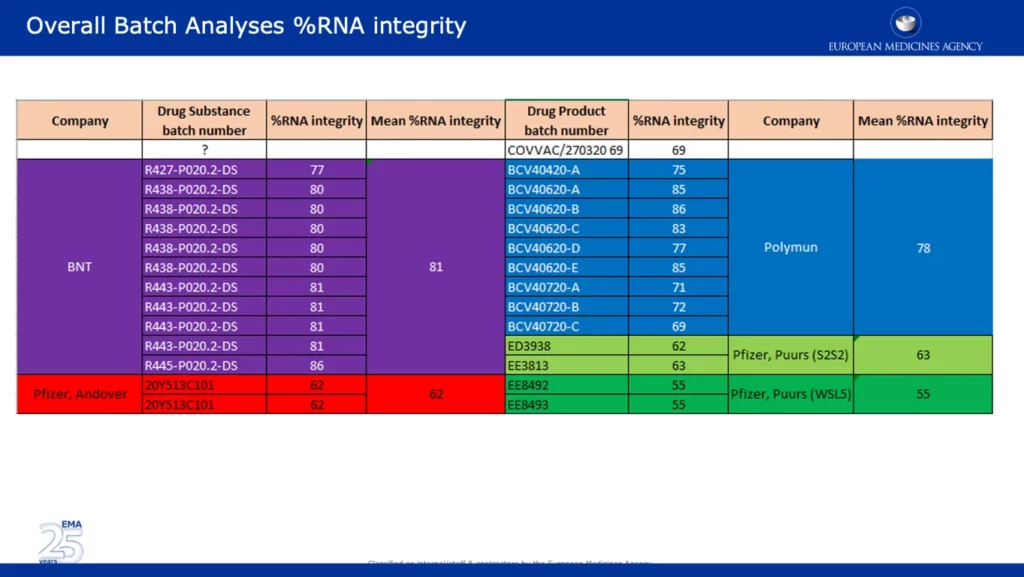
Skärmdumpen nedan är hämtad från den oredigerade APA som undertecknats av kommissionen. Där specificeras att majoriteten av Europas vaccinförsörjning kommer att ’komma från Pfizers tillverkningsanläggning i Puurs, Belgien.’ Detta är oroande med tanke på att uppgifterna visar att %RNA-integriteten för batcher från denna anläggning var den lägsta, 55 %, jämfört med andra anläggningar.

Bildtext:
1.6.3 Leveransmekanism
Vaccinleveranserna i Europa kommer huvudsakligen att komma från Pfizers tillverkningsanläggning i Puurs, Belgien, och ska innehålla RNA som producerats vid BioNTech-kontrollerade tillverkningsanläggningar, inklusive anläggningar som drivs av följande underleverantörer i Tyskland: (Slut på bildtext)
I skärmdumpen nedan visas godkännandekriterierna för olika BNT 162B2 läkemedelspartier. Med läkemedelsprodukt avses ’läkemedel i dess marknadsförda form, inklusive dess fyllnadsmedel, färgämnen och andra aktiva eller inaktiva substanser.’ Acceptanskriterierna för Emergency Supply är större än eller lika med 50 %, vilket råkar ligga strax under de partier med lägst %RNA-integritet som levererats av Pfizer, Puurs. Av någon anledning skiljer sig acceptanskriterierna för de kliniska läkemedelspartierna och sätts högre till större än eller lika med 60%.

CMC (Chemistry, Manufacturing and Controls) ’innebär att man definierar tillverkningsmetoder och produktspecifikationer som måste följas och uppfyllas för att garantera produktsäkerhet och överensstämmelse mellan satser.’ Med tanke på de data som visas ovan är det uppenbart att det fanns betydande CMC-problem när det gäller inkonsekvensen i %RNA-integriteten mellan batcher, vilket återspeglas i de läckta EMA-e-postmeddelandena och dokumenten. Det som är oroande är att tillverkaren (Pfizer/BioNTech) hävdade: ”Läkemedelsproduktens effekt är beroende av uttrycket av det levererade RNA, vilket kräver en tillräckligt intakt RNA-molekyl.”
Det är förbryllande hur man genom att sänka specifikationen till 50% kunde ’lösa’ denna viktiga invändning. Enligt en läckt Rapporteur’s Rolling Review Assessment (reviderat rapportdatum: 25 november 2020) som sammanställts av CHMP- och PRAC-rapportörerna – fann de de ’nuvarande acceptanskriterierna’ särskilt oroande. I rapporten anges att ’inga karakteriseringsdata om RNA-integritet och trunkerade former presenteras och de potentiella säkerhetsrisker som är förknippade med trunkerade RNA-isoformer tas inte upp. Detta är särskilt viktigt med tanke på att de nuvarande DS- och DP-godkännandekriterierna tillåter upp till 50 fragmenterade arter.’

Bildtext:
Förutom dubbelsträngat RNA finns det fler produktrelaterade föroreningar, t.ex. trunkerat RNA, även kallat fragmenterade arter. Trunkerat RNA återspeglas i DS-specifikationen när det gäller RNA-integritet. Karaktäriseringen av BNT162b2 DS anses dock för närvarande inte vara acceptabel i förhållande till CQA RNA-integritet. Betydande skillnader mellan satser tillverkade med process 1 och 2 observeras för detta specifika attribut. Även om två metoder, nämligen agarosgelelektrofores och kapillärgelelektrofores, har använts för att fastställa RNA-integriteten hos BNT162b2 DS, presenteras inga karakteriseringsdata om RNA-integritet och trunkerade former och de potentiella säkerhetsrisker som är förknippade med trunkerade RNA-isoformer tas inte upp. Detta är särskilt viktigt med tanke på att de nuvarande kriterierna för godkännande av DS och DP tillåter upp till 50% fragmenterade arter. Dokumentationen bör därför uppdateras med ytterligare karakteriseringsdata och diskussion om mRNA-integritet, vilket anses vara en viktig invändning. (Slut på bildtext)
Trunkerat (förkortat genom att antingen den översta eller den sista delen saknas) RNA definieras som en ’produktrelaterad orenhet’ och det faktum att potentiella säkerhetsrisker som härrör från denna fragmenterade art ’inte tas upp’ är mycket alarmerande.
Man hänvisar till ’de nuvarande acceptanskriterierna för DS och DP’, vilket innebär att de inte alltid har legat på den nivån och att de kanske har ändrats (sänkts). Frågan är varför? Kan det ha varit så att process 2 (tillverkningen av de kommersiella batcherna) helt enkelt inte var replikerbar på samma specifikationsnivå som de kliniska batcherna (småskalig) i process 1, och att en lägre standard därför sattes för att CMA skulle kunna beviljas?
Trial Site News kommunicerade med EMA angående innehållet i de läckta e-post-meddelandena och dokumenten. EMA:s presskontors snabba svar har publicerats nedan i sin helhet.
”Undersökningen av det publicerade materialet visade att korrespondensen manipulerades av förövarna före publiceringen. Inte alla dokument publicerades i sin fullständiga, ursprungliga form och kan ha tagits ur sitt sammanhang. Även om enskilda e-postmeddelanden var autentiska, valdes data från olika användare ut och sammanställdes, skärmdumpar från flera mappar och brevlådor skapades och ytterligare titlar lades till av förövarna.
Dessa dokument ger inte en fullständig bild av bedömningen av Comirnaty, covid-19-vaccinet som utvecklats av BioNTech/Pfizer. De visar situationen fram till början av december 2020, då knepet upptäcktes, men nämner inte den betydande mängd ytterligare data, information och förtydliganden som BioNTech/Pfizer lämnade in fram till den 21 december 2020, den dag då EMA:s kommitté för humanläkemedel (CHMP) gav sin rekommendation att bevilja ett godkännande för försäljning av detta vaccin.
Comirnaty verkar genom att det mRNA som det innehåller ger instruktioner för produktion av ett spikprotein som utlöser ett immunsvar. Dess effekt beror därför på förekomsten av en lämplig mängd intakt mRNA, som är känt för att vara relativt instabilt. Vad dokumenten visar är hur bedömningen av alla läkemedel går till: efter att ha granskat de uppgifter som företaget lämnat in hade CHMP frågor om mRNA:s integritet och tog upp dem formellt som en ’större invändning’. Detta är en integrerad del av bedömningen av alla läkemedel. Om viktiga invändningar förblir olösta hindrar de att godkännandet för försäljning beviljas. I detta fall hanterade företaget de frågor som tagits upp på ett tillfredsställande sätt och lämnade därefter in den information och de uppgifter som krävdes efter början av december 2020, vilket gjorde det möjligt för EMA att gå mot ett positivt yttrande för detta vaccin.
Det offentliga utredningsprotokollet för Comirnaty sammanfattar CHMP:s slutsatser i denna fråga och beskriver de steg som tagits under förfarandet för godkännande för försäljning av Comirnaty samt de skyldigheter som innehavaren av godkännandet för försäljning har att genomföra ytterligare studier för att noggrant övervaka den farmaceutiska kvaliteten på vaccinet. Dessa skyldigheter ingick också i den produktinformation som offentliggjordes vid tidpunkten för CHMP:s yttrande.
Även i en folkhälsokris som covid-19 har det alltid funnits en samsyn i EU om att inte kompromissa med standarder och att basera alla rekommendationer på tillgängliga vetenskapliga belägg för ett vaccins säkerhet, farmaceutisk kvalitet och effekt, och inget annat. Tillstånd beviljas endast när det finns övertygande bevis för att fördelarna med vaccination är större än de eventuella risker som ett vaccin medför.”
Artikeln publicerades ursprungligen i Trial Site News.
Suggest a correction