

A Further Investigation into the Leaked EMA Emails & Confidential Pfizer-BioNTech COVID-19 Vaccine Related Docs
In June, Trial Site News published a bombshell investigative report on the leaked European Medicine Agency (EMA) emails and other Pfizer-related confidential reports, which exposed concerning facts in the run-up to the authorization of the Pfizer-BioNTech COVID-19 vaccine. It revealed:
- A politically driven race between key regulators in their rush to authorize the vaccine.
- By late November 2020, regulators including, US FDA, European Medicines Agency, Health Canada and the UK’s MHRA, were all aware of the significant loss of RNA integrity of the commercial batches (~55% mRNA integrity) of the Pfizer-BioNTech vaccine compared to the clinical ones (~78% mRNA integrity). This was classified by EMA as a “major objection” along with observed visible particles, which were classified as “impurities.”
- A leaked 26 November PowerPoint presentation of a meeting between Pfizer-BioNTech and the EMA revealed how this major objection was shockingly ‘resolved’- the RNA integrity specification was simply lowered to 50%, therefore half of all mRNA molecules in the commercial batches were allowed to be truncated (not intact).
- The potential implications of the RNA integrity loss in terms of safety and efficacy were unknown.
This report focuses on further leaked emails with specific reference made to the European Commissioner, Ursula von der Leyen and the unusual extent she was willing to go in corralling member states (MSs) in avoiding the use of Article 5 (2) (their national Emergency Use Authorization for the COVID-19 vaccines) but go with an EU Conditional Marketing Authorization (CMA). It shines light on other leaked EMA sensitive documents: the 24 November, 2020 Quality Office CMC observations presentation by the BWP (Biologics Working party) and Rapporteur’s Rolling Review assessment report, revealing more evidence which supports the ‘major objections’ discussed in the original Trial Site News report. It looks at unredacted versions of Pfizer vaccine contracts and the EC’s Advance Purchase Agreement signed in November 2020 with Pfizer and BioNTech ‘for the development, production, priority-purchasing options and supply of a successful COVID-19 vaccine for EU Member States.’
No stranger to scandal
Ursula von der Leyen has been under intense public scrutiny over her private negotiations of a multi-billion-euro vaccine deal (the EU’s biggest contract) via secret texts and phone calls made with Pfizer’s CEO, Albert Bourla, contravening the Commission decision for a steering board to ‘provide guidance throughout the evaluation process.’ Furthermore, she refused to grant public access to the secretive text messages exchanged between Bourla and herself, when called to do so. A European Court of Auditors published an alarming report, which stated ‘we asked the Commission to provide us with information on the preliminary negotiations for this agreement (scientific experts consulted and advice received, timing of the talks, records of the discussions, and details of the agreed terms and conditions). However, none was forthcoming.’
The fire has also been heating up for Pfizer’s Bourla, when he was called to testify before the European Parliament’s Special Committee on COVID-19 to face questions over those secretive vaccine deals but last minute pulled out from facing the committee.
The leaked emails
The original Trial Site News investigative report highlighted the enormous pressure exerted by the EC (European Commission) on the EMA to grant CMA under a highly accelerated timeline. An email from Noel Wathion (EMA’s former deputy executive director) was published, which revealed a ‘rather tense’ TC (teleconference call) with the European Commissioner (Ursula von der Leyen) which was ‘at times even a bit unpleasant’.
‘A delay of several weeks..was not easily acceptable for the EC [European Commission]’ Wathion states. He also reveals ‘how political fall-out seems to be too high even if the “technical” level at the MSs could defend such a “delay” in order to make the outcome of the scientific review as robust as possible.’
Below is an email from Hilde Boone of the EMA, stating that ‘she [von der Leyen] will be prepared to call relevant health ministers personally to avoid the use of Art 5(2).’

Article 5 (2) vs CMA
Article 5 (2) of Directive 2001/83 states ‘Member States may temporarily authorise the distribution of an unauthorised medicinal product in response to the suspected or confirmed spread of pathogenic agents, toxins, chemical agents or nuclear radiation any of which could cause harm.’ In other words, it’s the equivalent of an Emergency Use Authorization at the member state level. It can be done very quickly because the medicinal product doesn’t need to go through the standard national authorization process.
An expert on EU law explained to Trial Site News the downside of MSs using Art. 5 (2), is that it would have given rise to competition amongst member states, leading to an unequitable access/distribution of the COVID-19 vaccines, particularly for those MSs who preferred to wait for the EU’s CMA (which takes longer since it’s supposed to follow a controlled and robust framework providing safeguards). An EU CMA helped ensure that the vaccines would have been available to all members states, at the same time, which was one of the objectives of the EU COVID-19 strategy.
However, could there have been further reasons why von der Leyen was desperate for the MSs to avoid Art 5 (2) that she was willing to call every relevant health minister herself?
In the email below from Noel Wathion, he states ‘since the 1st option still is to aim for a CMA rather than an Art 5 (2) coupled or not with an Art 5 (3)..’

Article 5(3) states: ‘Member States shall lay down provisions in order to ensure that marketing authorisation holders, manufacturers and health professionals are not subject to civil or administrative liability for any consequences resulting from the use of a medicinal product…’
The fact that Wathion points out that an Art. 5 (2) can be coupled or not with an Art. 5 (3), raises the important question whether member states would have had the option to not give indemnity to the marketing authorisation holders (in this case BioNTech) and manufacturers (BioNTech and Pfizer).
The Predatory Pfizer Contracts and Advance Purchase Agreements
Given what we know about the leaked Pfizer contracts made available by the non-profit consumer advocacy organization, Public Citizen; this pharmaceutical company has seemingly bullied countries into silence; can go after sovereign state assets (under the waiver of sovereign immunity) and enjoy full indemnity- exemption from legal liability that may result from their product- in fact it’s the Purchaser who is made liable on their behalf. This means governments have had to pay compensation to citizens who have suffered from a vaccine adverse event, not the vaccine manufacturer.
The screenshot below is taken from the unredacted version of the advance purchase agreement (APA) between the European Commission (EC), acting on behalf and in the name of member states, Pfizer Inc. and BioNTech (collectively ‘the Contractor’) signed in November 2020 (around the same time the leaked EMA emails were generated). It states ‘each Participating Member State shall indemnify and hold harmless the Contractor, their Affiliates..from and against any and all liabilities incurred..relating to harm, damages and losses..arising from or relating to the use and deployment of the Vaccines..’

However, information posted on the EC’s website states that under an EU Conditional Marketing Authorisation (CMA), ‘liability is with the holder of the marketing authorisation’ (which in this case is BioNTech). This directly conflicts with the APA that the EC signed with Pfizer and BioNTech (for 200 million vaccine doses priced at 15.50 Euros per dose excl VAT), a month before CMA was granted. It’s noteworthy that the EC published a heavily redacted version of the identical APA, any section related to indemnity/liability or simply considered ‘sensitive’ has been censored.
Another important point to consider when it comes to Art 5 (2) vs an EU CMA- is that given some European countries mandated the vaccine for adults, at-risk age groups and certain job sectors – it’s highly unlikely those MSs would have been able to do that with an unauthorised medicinal product, which these vaccines would have been classified under Art 5 (2).
The CMC issues
Wathion’s November 20, 2020 email raises several concerning points -‘there are still issues with both (CMC seems to be a concern for the Pfizer/BioNTech and the rolling review for Moderna just started giving less time to review) so it needs to be seen if all this can be sorted out on time, whilst not compromising the robustness of the review.’
The original Trial Site News report discussed what those CMC (Chemistry Manufacturing and Controls) issues were with Pfizer/BioNTech, particularly the loss of RNA integrity in the commercial batches and the unknown visible particles observed. Wathion tellingly states, ‘there are still issues’ speaks to the notion these ‘issues’ were not solved but had been ongoing. Wathion’s concern of ‘compromising the robustness of the review’ because of the need to authorise ‘on time’ is emphasized. This worry of speed over safety is reflected in other leaked emails, particularly by Wathion.
The report also contained a leaked email from Veronika Jekerle, EMA’s Head of Pharmaceutical Quality Office, where she outlined the 3 agreed major objections and the conclusion of the BWP (Biologics Working Party) regarding the Pfizer-BioNTech vaccine. Her email was sent on the November 24th the same day as the BWP presentation. Below, are a series of screenshots of the actual leaked BWP power point presentation (which Jekerle references in her email), entitled ‘EMA Quality Office CMC Observations.’ At the end of her email she thanks, ‘Ton, Brian and Claudio.’ Ton van der Stappen and Brian Dooley are named in the screenshot of the slide below.

The screenshot below presents wide-ranging data, which backups one of the major EMA objections in reference to the significant drop in %RNA integrity between the clinical and commercial batches. It’s noteworthy that batches from Pfizer’s Andover, USA (~62%) and Puurs, Belgium (~55%) sites were reported as having significantly lower %RNA integrity than batches provided by BNT (BioNTech) and Polymun.
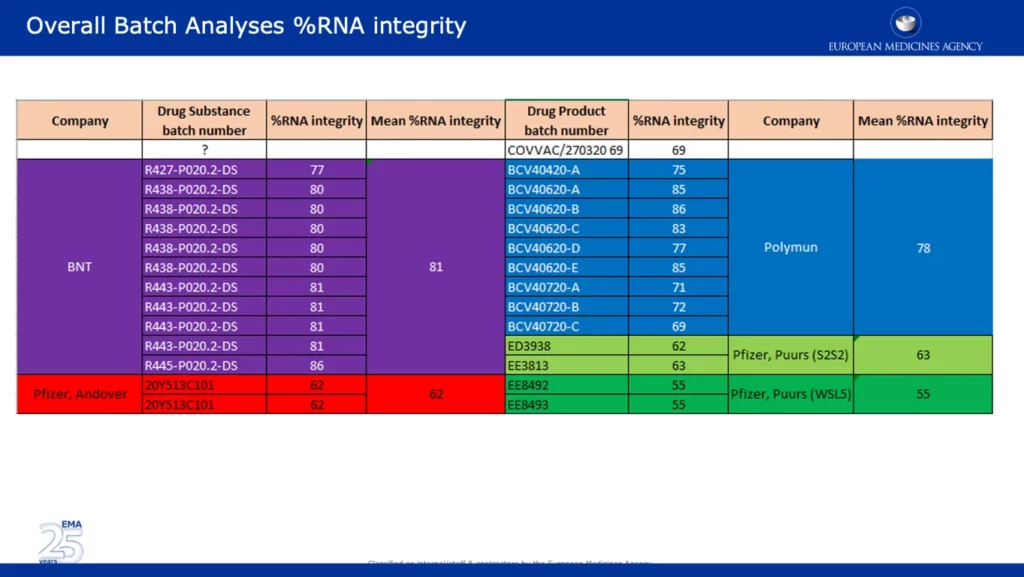
The screenshot below is taken from the unredacted APA signed by the EC. It specifies that the majority of Europe’s vaccine supply will ‘come from Pfizer’s manufacturing site in Puurs, Belgium.’ This is concerning given the data shows that the %RNA integrity of batches from that site were the lowest at 55%, compared to other sites.

The screenshot below shows the acceptance criteria of various BNT 162B2 Drug Product lots. Drug Product refers to ‘medication in its marketed form, including its fillers, coloring agents, and other active or inactive agents.’ The acceptance criteria for Emergency Supply is greater or equal to 50%, which happens to be just below the batches with the lowest %RNA integrity supplied by Pfizer, Puurs. For some reason the acceptance criteria for the clinical Drug Product Lots differs and is set higher at greater or equal to 60%.

CMC (Chemistry, Manufacturing and Controls) ‘involves defining manufacturing practices and product specifications that must be followed and met in order to ensure product safety and consistency between batches.’ Given the data shown above, it’s evident there were significant CMC issues regarding the inconsistency of the %RNA integrity between batches, which is reflected in the leaked EMA emails and documents. What’s concerning is that the manufacturer (Pfizer/BioNTech) claimed, “The efficacy of the drug product is dependent on the expression of the delivered RNA, which requires a sufficiently intact RNA molecule.”
It’s baffling how lowering the specification down to 50% was potentially how this major objection was ‘solved.’ According to a leaked Rapporteur’s Rolling Review Assessment (revised report date: 25 November, 2020) compiled by the CHMP and PRAC rapporteurs- they found the ‘current acceptance criteria’ particularly troubling. The report states, ‘no characterisation data on RNA integrity and truncated forms is presented and the potential safety risks associated with truncated RNA isoforms are not addressed. This is especially important considering that the current DS and DP acceptance criteria allows for up to 50% fragmented species.’

Truncated (shortened by missing either top or end section) RNA is defined as a ‘product-related impurity’ and the fact that potential safety risks arising from this fragmented species ‘are not addressed’ is highly alarming.
Reference is made to ‘the current DS and DP acceptance criteria,’ this implies that it was not always set at that level and perhaps it had been changed (lowered). The question is why? Could it have been that process 2 (the manufacturing of the commercial batches) was just not replicable at the same specification level to the clinical batches (small scale) of process 1, therefore a lower standard was set in order for CMA to be granted?
Trial Site News communicated with the EMA regarding the content of the leaked emails and documents. The EMA press office’s prompt response has been published below in its entirety.
“The investigation of the published material revealed that the correspondence was manipulated by the perpetrators prior to publication. Not all of the documents were published in their integral, original form and may have been taken out of context. Whilst individual emails were authentic, data from different users were selected and aggregated, screenshots from multiple folders and mailboxes were created and additional titles were added by the perpetrators.
These documents do not present a full picture of the assessment of Comirnaty, the COVID-19 vaccine developed by BioNTech/Pfizer. They show the situation up to the beginning of December 2020, when the hack was discovered, but do not mention the considerable amount of additional data, information and clarifications submitted by BioNTech/Pfizer up to 21 December 2020, the day when EMA’s committee for human medicines (CHMP) gave its recommendation to grant a marketing authorization for this vaccine.
Comirnaty works because the mRNA it contains provides instructions for producing a spike protein which triggers an immune response. Its efficacy therefore depends on the presence of suitable amount of intact mRNA, which is known to be relatively unstable. What the documents show is how the assessment of any medicine works: following scrutiny of the data submitted by the company, the CHMP had questions about the integrity of mRNA and raised them formally as a ‘major objection’. This is an integral part of the assessment of any medicine. If major objections remain unresolved, they preclude the granting of the marketing authorisation. In this case, the company addressed the issues raised satisfactorily and subsequently supplied the required information and data after beginning of December 2020, which allowed EMA to move towards a positive opinion for this vaccine.
The public assessment report of Comirnaty summarises the conclusions of the CHMP on this issue and details the steps taken during the marketing authorization procedure of Comirnaty as well as the obligations placed on the marketing authorisation holder to carry out additional studies to closely monitor the pharmaceutical quality of vaccine. These obligations were also included in the Product Information published at the time of the CHMP opinion.
Even in a public health emergency as COVID-19, there has always been consensus across the EU not to compromise standards and to base any recommendation on the available scientific evidence on a vaccine’s safety, pharmaceutical quality and efficacy, and nothing else. Authorizations are only granted when the evidence shows convincingly that the benefits of vaccination are greater than any risks posed by a vaccine.”
Originally published in Trial Site News.
Suggest a correction




