Suite de l’enquête et révélations des emails de l’EMA et des documents confidentiels de Pfizer-BioNTech relatifs au vaccin COVID-19
par Sonia Elijah
En juin, Trial Site News a publié un rapport d’enquête retentissant sur la fuite de courriels de l’Agence européenne du médicament (EMA) et d’autres rapports confidentiels liés à Pfizer, qui mettaient en lumière des faits relatifs à la préparation de l’autorisation du vaccin COVID-19 de Pfizer-BioNTech. Le rapport a révélé :
- Une course politique entre les principaux régulateurs dans leur empressement à autoriser le vaccin.
- Fin novembre 2020, les organismes de réglementation, dont la FDA américaine, l’Agence européenne des médicaments, Santé Canada et la MHRA britannique, étaient tous au courant de la perte significative de l’intégrité de l’ARN des lots commerciaux (~55% d’intégrité de l’ARNm) du vaccin Pfizer-BioNTech par rapport aux lots cliniques (~78% d’intégrité de l’ARNm). Cela a été classé par l’EMA comme une « objection majeure », tout comme les particules visibles observées, qui ont été classées comme « impuretés ».
- Une présentation PowerPoint d’une réunion entre Pfizer-BioNTech et l’EMA, qui a fait l’objet d’une fuite le 26 novembre, a révélé comment cette objection majeure a été « résolue » de manière choquante – la spécification d’intégrité de l’ARN a simplement été abaissée à 50 %, ce qui signifie que la moitié de toutes les molécules d’ARNm dans les lots commerciaux ont été autorisées à être tronquées (non intactes).
- Les implications potentielles de la perte d’intégrité de l’ARN en termes de sécurité et d’efficacité étaient inconnues.
- Le présent rapport se concentre sur d’autres courriels ayant fait l’objet d’une fuite et fait spécifiquement référence à la commissaire européenne Ursula von der Leyen et à la mesure inhabituelle qu’elle était prête à prendre pour inciter les États membres à ne pas recourir à l’article 5, paragraphe 2 (leur autorisation nationale d’utilisation d’urgence pour les vaccins COVID-19) mais à opter pour une autorisation de mise sur le marché conditionnelle (AMC). Il met en lumière d’autres documents sensibles de l’EMA qui ont fait l’objet d’une fuite : la présentation des observations CMC de l’Office de la qualité du 24 novembre 2020 par le BWP (Biologics Working Party) et le rapport d’évaluation de l’examen glissant du rapporteur, révélant davantage de preuves qui soutiennent les « objections majeures » discutées dans le rapport original de Trial Site News. Il examine les versions non expurgées des contrats de vaccins de Pfizer et l’accord d’achat anticipé de la CE signé en novembre 2020 avec Pfizer et BioNTech « pour le développement, la production, les options d’achat prioritaire et la fourniture d’un vaccin COVID-19 réussi pour les États membres de l’UE ».
Habituée au scandale
Ursula von der Leyen a fait l’objet d’un examen public intense concernant ses négociations privées d’un contrat de vaccins de plusieurs milliards d’euros (le plus gros contrat de l’UE) par le biais de textos et d’appels téléphoniques secrets avec le PDG de Pfizer, Albert Bourla, en violation de la décision de la Commission de mettre en place un comité directeur chargé de « fournir des orientations tout au long du processus d’évaluation« . En outre, elle a refusé d’accorder au public l’accès aux textos secrets échangés entre Bourla et elle-même, alors qu’elle y était invitée. La Cour des comptes européenne a publié un rapport alarmant selon lequel « nous avons demandé à la Commission de nous fournir des informations sur les négociations préliminaires de cet accord (experts scientifiques consultés et avis reçus, calendrier des discussions, comptes rendus des discussions et détails des conditions convenues). Cependant, aucune information n’a été fournie.«
Le feu s’est également échauffé pour Bourla de Pfizer, lorsqu’il a été appelé à témoigner devant la commission spéciale du Parlement européen sur le COVID-19 pour répondre aux questions concernant ces accords secrets sur les vaccins, mais il s’est retiré à la dernière minute de la commission.
Fuite de courriels
Le rapport d’enquête original de Trial Site News a mis en évidence l’énorme pression exercée par la CE (Commission européenne) sur l’EMA pour qu’elle accorde l’AMC dans un délai très accéléré. Un courriel de Noel Wathion (l’ancien directeur exécutif adjoint de l’EMA) a été publié, révélant une téléconférence « plutôt tendue » avec la commissaire européenne (Ursula von der Leyen) qui était « parfois même un peu désagréable« .
« Un retard de plusieurs semaines… était difficilement acceptable pour la CE [Commission européenne]« , déclare Wathion. Il révèle également « comment les retombées politiques semblent être trop importantes, même si le niveau « technique » des États membres pourrait défendre un tel « retard » afin de rendre le résultat de l’examen scientifique aussi solide que possible« .
Vous trouverez ci-dessous un courriel de Hilde Boone de l’EMA, indiquant qu' »elle [von der Leyen] sera prête à appeler personnellement les ministres de la santé concernés pour éviter le recours à l’article 5, paragraphe 2« .

Article 5, paragraphe 2, contre CMA
L’article 5, paragraphe 2, de la directive 2001/83 stipule que « les États membres peuvent autoriser temporairement la distribution d’un médicament non autorisé en réponse à la propagation suspectée ou confirmée d’agents pathogènes, de toxines, d’agents chimiques ou de radiations nucléaires susceptibles de causer des dommages« . En d’autres termes, c’est l’équivalent d’une autorisation d’utilisation d’urgence au niveau des États membres. Elle peut être accordée très rapidement, car le médicament n’a pas besoin de passer par le processus d’autorisation national standard.
Un expert en droit européen a expliqué à Trial Site News que l’inconvénient pour les États membres d’utiliser l’article 5, paragraphe 2, est que cela aurait donné lieu à une concurrence entre les États membres, entraînant un accès/distribution inéquitable des vaccins COVID-19, en particulier pour les États membres qui ont préféré attendre l’AMC de l’UE (qui prend plus de temps puisqu’elle est censée suivre un cadre contrôlé et robuste fournissant des garanties). L’AMC de l’UE a permis de garantir que les vaccins auraient été disponibles pour tous les États membres, au même moment, ce qui était l’un des objectifs de la stratégie européenne COVID-19.
Cependant, y avait-il d’autres raisons pour lesquelles Mme von der Leyen souhaitait désespérément que les États membres évitent l’article 5, paragraphe 2, au point d’appeler elle-même tous les ministres de la santé concernés ?
Dans l’e-mail ci-dessous de Noel Wathion, il déclare « puisque la première option reste de viser une AMC plutôt qu’un article 5 (2) couplé ou non à un article 5 (3)…« .

L’article 5, paragraphe 3, dispose que « Les États membres prévoient des dispositions afin que les titulaires d’autorisations de mise sur le marché, les fabricants et les professionnels de la santé ne soient pas soumis à une responsabilité civile ou administrative pour toute conséquence résultant de l’utilisation d’un médicament…« .
Le fait que Wathion indique que l’article 5, paragraphe 2, peut être couplé ou non à l’article 5, paragraphe 3, soulève la question importante de savoir si les États membres auraient eu la possibilité de ne pas indemniser les titulaires d’une autorisation de mise sur le marché (dans ce cas, BioNTech) et les fabricants (BioNTech et Pfizer).
Les contrats prédateurs de Pfizer et les accords d’achat anticipé
Compte tenu de ce que nous savons des contrats de Pfizer qui ont fait l’objet de fuites et qui ont été rendus publics par l’organisation de défense des consommateurs à but non lucratif Public Citizen, il semble que cette société pharmaceutique ait contraint des pays au silence, qu’elle peut s’en prendre aux droits d’États souverains (en vertu de la renonciation à l’immunité souveraine) et qu’elle bénéficie d’une indemnisation totale, c’est-à-dire d’une exemption de toute responsabilité juridique pouvant résulter de son produit – en fait, c’est l’acheteur qui est tenu responsable en leur nom. Cela signifie que les gouvernements ont dû verser des indemnités aux citoyens qui ont souffert de l’effet indésirable d’un vaccin, et non le fabricant du vaccin.
La capture d’écran ci-dessous est tirée de la version non expurgée de l’accord d’achat anticipé (APA) entre la Commission européenne (CE), agissant au nom et pour le compte des États membres, Pfizer Inc. et BioNTech (collectivement « le contractant« ), signé en novembre 2020 (à peu près au moment où les courriels de l’EMA ont été générés). Il stipule que « chaque État membre participant indemnisera et dégagera de toute responsabilité le contractant, ses affiliés… pour toute responsabilité encourue… liée à des préjudices, dommages et pertes… découlant de ou liés à l’utilisation et au déploiement des vaccins…« .

Cependant, les informations publiées sur le site web de la CE indiquent que dans le cadre d’une autorisation de mise sur le marché conditionnelle (AMC) de l’UE, « la responsabilité incombe au titulaire de l’autorisation de mise sur le marché » (qui, dans ce cas, est BioNTech). Ceci est en contradiction directe avec l’APA que la CE a signé avec Pfizer et BioNTech (pour 200 millions de doses de vaccin au prix de 15,50 euros par dose hors TVA), un mois avant que l’AMC ne soit accordée. Il convient de noter que la CE a publié une version fortement expurgée de l’APP identique, toute section relative à l’indemnisation/la responsabilité ou simplement considérée comme « sensible » ayant été censurée.
Un autre point important à considérer en ce qui concerne l’article 5 (2) par rapport à une AMC de l’UE est que, étant donné que certains pays européens ont rendu le vaccin obligatoire pour les adultes, les groupes d’âge à risque et certains secteurs d’emploi, il est très peu probable que ces États membres aient pu le faire avec un produit médicinal non autorisé, ce que ces vaccins auraient été classés sous l’article 5 (2).
Les enjeux du CMC
L’e-mail de Wathion du 20 novembre 2020 soulève plusieurs points préoccupants – « il y a encore des problèmes avec les deux (la CMC semble être une préoccupation pour Pfizer/BioNTech et la révision continue pour Moderna vient de commencer, ce qui donne moins de temps pour la révision), il faut donc voir si tout cela peut être réglé à temps, sans compromettre la robustesse de la révision« .
Le rapport original de Trial Site News abordait les problèmes de CMC (chimie, fabrication et contrôles) rencontrés par Pfizer/BioNTech, notamment la perte d’intégrité de l’ARN dans les lots commerciaux et les particules visibles inconnues observées. La déclaration éloquente de Wathion, « il y a encore des problèmes « , laisse entendre que ces « problèmes » n’étaient pas résolus mais qu’ils étaient en cours. La crainte de Wathion de « compromettre la robustesse de l’examen » en raison de la nécessité d’autoriser « à temps » est soulignée. Cette préoccupation de la rapidité au détriment de la sécurité se reflète dans d’autres courriels ayant fait l’objet d’une fuite, notamment ceux de Wathion.
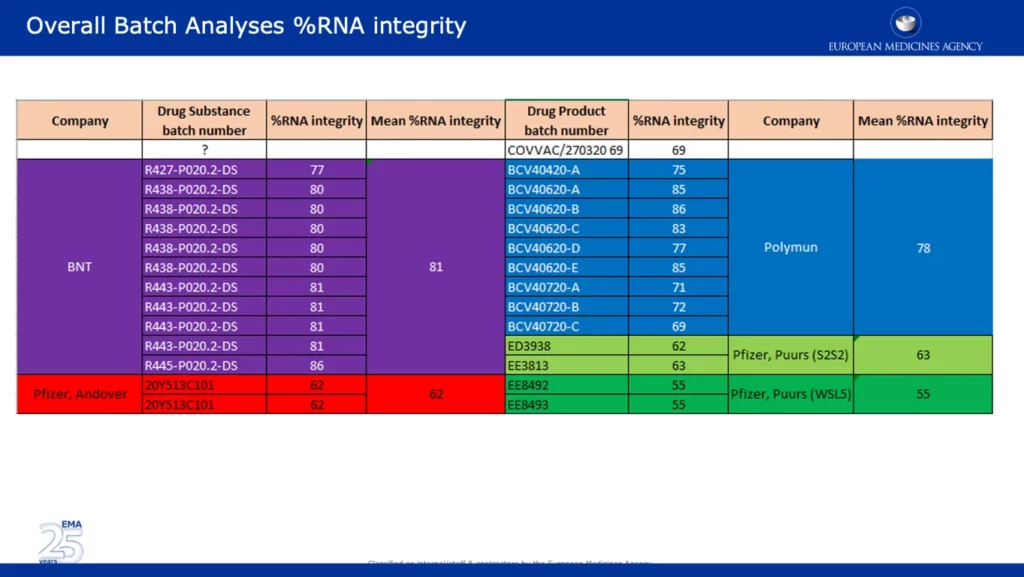
Le rapport contient également un courriel de Veronika Jekerle, chef du Bureau de la qualité pharmaceutique de l’EMA, dans lequel elle expose les trois objections majeures retenues et la conclusion du BWP (Biologics Working Party) concernant le vaccin Pfizer-BioNTech. Son courriel a été envoyé le 24 novembre, le même jour que la présentation du BWP. Vous trouverez ci-dessous une série de captures d’écran de la présentation PowerPoint du BWP qui a fait l’objet d’une fuite (à laquelle Jekerle fait référence dans son courriel), intitulée « EMA Quality Office CMC Observations« . À la fin de son courriel, elle remercie « Ton, Brian et Claudio« . Ton van der Stappen et Brian Dooley sont nommés dans la capture d’écran de la diapositive ci-dessous.

La capture d’écran ci-dessous présente des données très variées, qui confirment l’une des principales objections de l’EMA concernant la baisse significative du % d’intégrité de l’ARN entre les lots cliniques et commerciaux. Il est intéressant de noter que les lots provenant des sites de Pfizer à Andover, aux États-Unis (~62%) et à Puurs, en Belgique (~55%) ont été signalés comme ayant un % d’intégrité d’ARN significativement plus faible que les lots fournis par BNT (BioNTech) et Polymun.
La capture d’écran ci-dessous est tirée de l’APP non expurgé signé par la CE. Il précise que la majorité de l’approvisionnement en vaccins de l’Europe « proviendra du site de fabrication de Pfizer à Puurs, en Belgique« . Ceci est inquiétant étant donné que les données montrent que le % d’intégrité de l’ARN des lots provenant de ce site était le plus bas, à 55%, par rapport aux autres sites.

La capture d’écran ci-dessous montre les critères d’acceptation de divers lots de produits pharmaceutiques BNT 162B2. Le terme « produit pharmaceutique » désigne « le médicament sous sa forme commercialisée, y compris les agents de remplissage, les colorants et les autres agents actifs ou inactifs« . Le critère d’acceptation pour l’approvisionnement d’urgence est supérieur ou égal à 50 %, ce qui se trouve être juste en dessous des lots présentant le plus faible pourcentage d’intégrité de l’ARN fourni par Pfizer, Puurs. Pour une raison quelconque, le critère d’acceptation pour les lots de produits pharmaceutiques cliniques diffère et est fixé à un niveau supérieur ou égal à 60 %.

La CMC (chimie, fabrication et contrôles) « implique la définition des pratiques de fabrication et des spécifications du produit qui doivent être suivies et respectées afin de garantir la sécurité du produit et la cohérence entre les lots« . Au vu des données présentées ci-dessus, il est évident qu’il y avait d’importants problèmes de CMC concernant l’incohérence de l’intégrité du %RNA entre les lots, ce qui est reflété dans les courriels et documents de l’EMA qui ont fait l’objet d’une fuite. Ce qui est inquiétant, c’est que le fabricant (Pfizer/BioNTech) a affirmé que « l’efficacité du produit pharmaceutique dépend de l’expression de l’ARN délivré, ce qui nécessite une molécule d’ARN suffisamment intacte.«
Il est déconcertant de constater que c’est en abaissant la spécification à 50 % que cette objection majeure a pu être « résolue« . Selon une fuite de l’évaluation de l’examen glissant du rapporteur (date du rapport révisé : 25 novembre 2020) compilée par les rapporteurs du CHMP et du PRAC – ils ont trouvé les « critères d’acceptation actuels » particulièrement troublants. Selon le rapport, « aucune donnée de caractérisation sur l’intégrité de l’ARN et les formes tronquées n’est présentée et les risques potentiels de sécurité associés aux isoformes d’ARN tronquées ne sont pas abordés. Ceci est particulièrement important si l’on considère que les critères d’acceptation actuels des DS et DP autorisent jusqu’à 50% d’espèces fragmentées« .

L’ARN tronqué (raccourci par l’absence de la section supérieure ou de la section finale) est défini comme une « impureté liée au produit » et le fait que les risques de sécurité potentiels découlant de cette espèce fragmentée « ne sont pas abordés » est très alarmant.
Il est fait référence aux « critères d’acceptation DS et DP actuels« , ce qui implique qu’ils n’ont pas toujours été fixés à ce niveau et qu’ils ont peut-être été modifiés (abaissés). La question est de savoir pourquoi ? Se pourrait-il que le processus 2 (la fabrication des lots commerciaux) ne soit tout simplement pas reproductible au même niveau de spécification que les lots cliniques (à petite échelle) du processus 1, et qu’une norme inférieure ait donc été fixée pour que l’AMC soit accordée ?
Trial Site News a communiqué avec l’EMA au sujet du contenu des courriels et documents ayant fait l’objet d’une fuite. La réponse rapide du bureau de presse de l’EMA a été publiée ci-dessous dans son intégralité.
« L’enquête sur les documents publiés a révélé que la correspondance a été manipulée par les auteurs avant la publication. Tous les documents n’ont pas été publiés dans leur forme intégrale et originale et peuvent avoir été sortis de leur contexte. Alors que les courriels individuels étaient authentiques, les données de différents utilisateurs ont été sélectionnées et agrégées, des captures d’écran de plusieurs dossiers et boîtes aux lettres ont été créées et des titres supplémentaires ont été ajoutés par les auteurs.
Ces documents ne présentent pas une image complète de l’évaluation de Comirnaty, le vaccin COVID-19 développé par BioNTech/Pfizer. Ils montrent la situation jusqu’au début du mois de décembre 2020, lorsque le piratage a été découvert, mais ne mentionnent pas la quantité considérable de données, d’informations et de clarifications supplémentaires soumises par BioNTech/Pfizer jusqu’au 21 décembre 2020, jour où le comité des médicaments humains (CHMP) de l’EMA a donné sa recommandation d’accorder une autorisation de mise sur le marché pour ce vaccin.
Le Comirnaty fonctionne car l’ARNm qu’il contient fournit des instructions pour produire une protéine de pointe qui déclenche une réponse immunitaire. Son efficacité dépend donc de la présence d’une quantité adéquate d’ARNm intact, dont on sait qu’il est relativement instable. Les documents montrent comment fonctionne l’évaluation de tout médicament : après avoir examiné les données soumises par la société, le CHMP s’est posé des questions sur l’intégrité de l’ARNm et les a soulevées officiellement en tant qu' »objection majeure ». Cela fait partie intégrante de l’évaluation de tout médicament. Si les objections majeures ne sont pas résolues, elles empêchent l’octroi de l’autorisation de mise sur le marché. Dans le cas présent, la société a répondu de manière satisfaisante aux questions soulevées et a ensuite fourni les informations et données requises après début décembre 2020, ce qui a permis à l’EMA de s’orienter vers un avis positif pour ce vaccin.
Le rapport d’évaluation public de Comirnaty résume les conclusions du CHMP sur cette question et détaille les mesures prises au cours de la procédure d’autorisation de mise sur le marché de Comirnaty ainsi que les obligations imposées au titulaire de l’autorisation de mise sur le marché de réaliser des études supplémentaires pour surveiller étroitement la qualité pharmaceutique du vaccin. Ces obligations ont également été incluses dans l’information sur le produit publiée au moment de l’avis du CHMP.
Même dans une situation d’urgence de santé publique comme celle du COVID-19, il y a toujours eu un consensus au sein de l’UE pour ne pas compromettre les normes et pour fonder toute recommandation sur les preuves scientifiques disponibles concernant la sécurité, la qualité pharmaceutique et l’efficacité d’un vaccin, et rien d’autre. Les autorisations ne sont accordées que lorsque les preuves montrent de manière convaincante que les avantages de la vaccination sont supérieurs aux risques éventuels d’un vaccin.«
Publié originellement dans Trial Site News.
Suggérer une correction