What the Leaked EMA Emails & Docs Reveal: Major Concerns with Pfizer C-19 Vaccine Batch Integrity and The Race to Authorize
Our recent article by Meryl Nass, MD on the fastest vaccine roll out in world history detailing the approval procedure for the new mRNA ‘vaccines’, highlights the incomprehensible ignorance of how the health authorities function today.
At the same time, the European Parliament Special Committee on COVID-19 pandemic had the opportunity to pose questions to the representatives of four pharmaceutical companies on September 5th, 2022. The meeting focused on pandemic related activities of the past and future with a hopeless attempt by a few to bring transparency and accountability expected in a democratic setting. Mr. Cristian Terhes, European Parliament member has raised the right questions.
For anyone who may be shocked by the quick roll out of the new ‘vaccines’ in Europe, we must remember and question how the authorization of previous COVID-19 vaccination policy went, as we learnt from leaked emails from the EMA (European Medicines Agency), summarised in the following article by the investigative journalist and broadcaster, Sonia Elijah, on Trial Site News (June 2022).
What the Leaked EMA Emails & Docs Reveal: Major Concerns with Pfizer C-19 Vaccine Batch Integrity and The Race to Authorize
Trial Site News (video above) were recently able to review leaked internal emails from the European Medicines Agency (EMA) and meeting report between the agency and Pfizer. The EMA oversees the evaluation and supervision of medicinal products for the European Union. Like other regulatory health bodies, its main responsibility is to protect and promote public health. Snapshots of internal EMA email correspondence; a November 26, 2020, PowerPoint presentation from a pivotal meeting between Pfizer and the agency, as well as a confidential 43-page Pfizer report were provided by an anonymous source because of their trust in Trial Site’s commitment to transparency, accessibility, and accountability in furtherance of a highly ethical, quality-focused and public health-centric biomedical research industry.
Regulatory agencies, like the EMA, the Food and Drug Administration (FDA) in the U.S. and the UK’s Medicines and Healthcare products Regulatory Agency (MHRA) are chartered to make decisions based to better the public. External influences such as political or media pressure are not meant to be a driving factor in their decision-making, however, when it came to pandemic conditions and the fast-tracked conditional marketing authorization of the Covid-19 vaccines (particularly for the mRNA-based vaccines produced by Pfizer-BioNTech and Moderna), it appears the latter won the day.
The time period of the email correspondence in question stretches from November 10 – 25, 2020, just weeks before the EMA granted CMA (conditional marketing authorization) for the Pfizer-BioNTech Covdid-19 vaccine on December 21, 2020. The FDA granted EUA (emergency use authorization) for this vaccine on December 11 with the MHRA making it first to the finish line on December 2. Here this author uses the term ‘finish line,’ as the emails do reveal an intense, almost competitive-like rush to authorize the Covid-19 vaccines, as quickly as possible. Understandably, the world was gripped by a pandemic at the time, where there was immense impetus to authorize a vaccine to protect people from the novel coronavirus.
The Rush into EUA
In an email from Marco Cavaleri, at the time the EMA’s Head of Biological Health Threats and Vaccines Strategy, communicated with urgency how the U.S. FDA “are going to rush into EUA.”
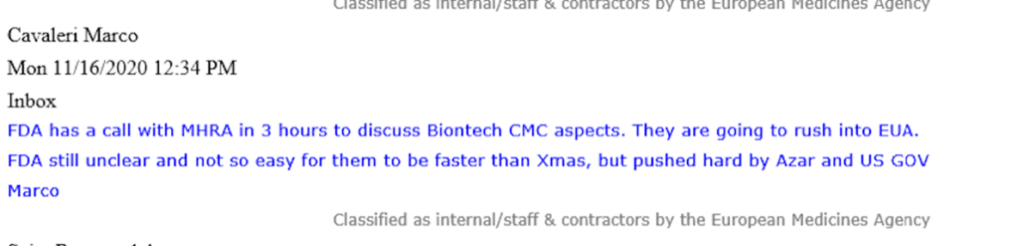
Cavaleri refers to this ‘rush’ being ‘pushed hard by Azar and US GOV.’ Under the Trump administration, Alex Azar, former pharmaceutical executive was the United States Secretary of Health and Human Services (HHS) from 2018-2021. The FDA is an agency that falls directly under the HHS.
It’s worth noting that when Azar was former president of Lilly USA LLC, a division of Eli Lilly, drug prices skyrocketed under his leadership. The pharmaceutical company was also embroiled in a class-action lawsuit under his tenure where it was accused of exploiting the drug pricing system to increase profits for its insulin drug. Of course, this doesn’t necessarily mean this executive was complicit in any way, but the timing is noteworthy.
Cavaleri’s email speaks to the extent of how politics (and the US government) was driving the FDA’s regulatory process, making sure it was going at ‘warp speed’. And of course, on that note Trump’s Operation Warp Speed was to ensure all vaccine development records would be shattered. The intentions were undoubtedly good given the outbreak of the worst pandemic in a century.
However, across the Atlantic in Europe’s regulatory agency tension mounted as the pressure to accelerate deadlines made the air and general mood tense—the pressure and anxiety was palpable in the reviewed email exchanges.
Persons of high integrity and clarity as to their roles and commitments as stewards of public health emerged. For example, one individual demonstrated palpable concern over accelerated timelines to ensure they would meet the ‘deadline’ for vaccine authorization at the expense of a robust assessment. He was Noel Wathion, at the time the EMA’s deputy executive director, but who has since retired. This EMA official importantly pointed out, ‘We are speeding up as much as possible, but we also need to make sure that our scientific assessment is as robust as possible. Let’s not forget the responsibility/accountability attached to the recommendation to the EC to grant a CMA.’

Wathion assumes the FDA (and the MHRA’s) EUA would be issued before the EMA granted its own CMA, which turned out to be correct. What’s interesting is his concern to address the ‘damage limitation’ resulting from the probable outcome of the EMA finishing last in this regulatory race and his fear that this would result in public opinion and the media turning against the agency. Speed seemingly superseded concerns of quality based on a careful review of these emails.
In a November 19 email, Wathion reveals a ‘rather tense’ TC (teleconference call) with the European Commissioner (Ursula von der Leyen) which was ‘at times even a bit unpleasant.’ This reflects the mounting pressure which the EMA staff were under to issue CMA quickly following an EUA granted by the FDA/MHRA for the Pfizer-BioNTech vaccine. Von der Leyen is implicated in potentially being responsible for this tense environment with ‘a delay of several weeks…not easily acceptable for the EC [European Commission].’
In early 2022, Trial Sites News reported how von der Leyen was embroiled in scandal when a group of independent MEPs demanded her immediate resignation and full disclosure of a series of private text messages between her and Pfizer’s CEO, Albert Bourla. Only a small portion of these texts were ever disclosed. Of the ones that were, they revealed her negotiating portions of a European-wide vaccine deal, unilaterally with Bourla via a series of texts! Clearly standard protocols in Europe were thrown out the window in favor of expediency and this seemingly was tied to a unified competitive pressure on all three regulatory agencies.
Wathion lays bare his reflections after this particular TC, and shockingly writes how ‘the political fall-out seems to be too high even if the “technical” level at the MSs [Member States] could defend such a delay in order to make the outcome of the scientific review as robust as possible.’ Put another way the continuous broadcast of science first appeared as a cover for politics first.

Wathion points out that a potential delay of several weeks to secure ‘robust assurance in particular as regards CMC and safety’ will be met with ‘criticism from various parties,’ including media, EC (European Commission) and EP (European Parliament). Wathion speaks of his fear that if the deadline ‘to align as much as possible with the “approval” timing by FDA/MHRA’ cannot be met- ‘we will be overwhelmed from all fronts and be in the middle of the storm.’ However, this potential delay appeared to be necessary ‘in order to make the outcome of the scientific review as robust as possible.’ This implies that speed at the expense of safety was the order of the day to avoid ‘political fallout.’ Clearly, politics was dictating Covid-19 vaccine authorisation protocol, not the science.

In the above email from Marco, the EMA official reveals that Pfizer’s CEO Albert Bourla ‘lobbied’ Peter Marks, and this could be interpreted as highly controversial, given Marks is the director of the Center for Biologics Evaluation and Research (CBER) at the FDA. Pfizer’s apparent access into the federal watchdog raises significant questions at the least, if not introduces the possibility for disturbing entanglements between industry and a purportedly independent, scientific federal agency.
Major concerns with the integrity between vaccine batches
An email from Cavaleri (see below) reveals at that time the FDA knew of ‘some issues’ associated with the CMC which needed to be sorted out and may ‘end up being the difficult bit.’ CMC refers to the Chemistry, Manufacturing and Controls, also referred to as pharmaceutical quality, which covers various procedures used to assess and ensure the safety and consistency between pharmaceutical product batches.
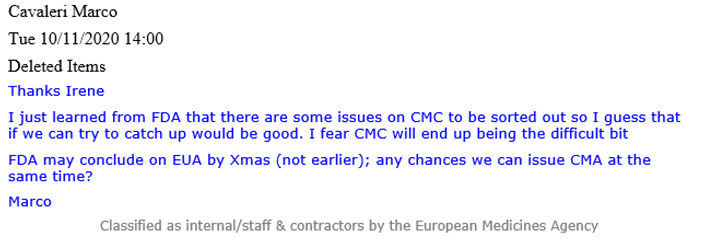
An email from Evdokia Korakianiti (an EMA scientific administrator) explains in more detail what these “issues” were and how they were in fact major concerns to do with the Pfizer- BioNTech vaccine.

Alarmingly, significant differences in the levels of mRNA integrity between Pfizer-BioNTech’s commercial (large scale) and clinical vaccine batches (small scale) were observed. ‘~78% mRNA integrity’ in the clinical ones and ‘~ 55% in the proposed commercial batches’ with the ‘root cause’ not yet identified. Safety and efficacy implications due to this concern were also noted in the email ‘as yet to be defined.’
In a confidential Pfizer report, which was also leaked along with the EMA emails, the company states that according to Acuitas Therapeutics’ (the biotech company who developed the lipid nanoparticle platform for the Pfizer and Moderna vaccine) own general experience, ‘a minimum threshold is approximately 70%.’ (See screenshot below)

Then on page 30 it states: ‘The efficacy of the product is dependent on expression of the delivered RNA, which requires a sufficiently intact RNA molecule.’ (See screenshot below)

This exact phrase ‘requires a sufficiently intact RNA molecule’ was used in the email from EMA staffer, Evdokia Korakianiti, which I included above, sent on November 23, 2020- now we likely know where Korakianiti referenced it from.
For the commercial batches (which were going to be rolled out across the globe) to have such a significantly lower level of mRNA integrity (intact RNA molecule) is greatly concerning given its intrinsic tie to the efficacy and potential safety of the product.
The next day Veronika Jekerle, Head of Pharmacy Quality Office, writes to Evdokia (see below).

The difference in the level of mRNA integrity was again noted as a major concern ‘shared by most member states’ and its ‘potential impact on safety.’ Jekerle highlights in bold, “An approval by the end of the year could potentially be possible, if these concerns + GMP will be resolved.”
This gives rise to the critical question- how were all these concerns resolved when CMA was granted only a few weeks later, on December 21? A possible way it was resolved is explained later in this report.
In contrast to the concerns of some of the other EMA officials, Marco Cavaleri writes around the same time in the following email (see below) that the mRNA content is not a major concern, according to the FDA- ‘the issue on the mRNA content not perceived as major.’ He also shockingly states, ‘unclear if GCP inspections ever done.’ This revelation is highly concerning given that GCP refers to Good Clinical Practise, which is ‘an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human subjects.’
What’s even more alarming is his following statement- ‘no major interest from FDA.’ This looks to reveal the regulatory agency’s apparent lack of concern or even interest on whether GCP inspections were completed, in the context of Pfizer’s clinical trials, which was relied on by the FDA to grant EUA for the Pfizer-BioNTech vaccine. In one of this author’s previous investigative reports for Trial Site News, we noted that the FDA only inspected 1% of Pfizer’s trial sites.

Further damming information is revealed (see screenshot below) when multiple regulatory agencies: Health Canada (HC), EMA, MHRA and FDA are all aware of the issue with % mRNA integrity, yet FDA and Health Canada make an unsubstantiated claim that ‘safety concerns associated. Are more of a theoretical concern.’

Health Canada then appears to contradict itself because its later described as showing particular concern about one region receiving ‘all the suboptimal material.’ Obviously, it didn’t want to be that region.
Shockingly, the end of the email reveals the ‘Applicant [Pfizer] has shared with FDA and us [EMA]/MHRA only today and issue with visible particles in the DP [drug product] appears to be lipid nanoparticle components.)’
This is highly concerning due to this significant issue being made known to the three key regulatory agencies on November 25, only a few weeks away before the EMA granted CMA and the FDA granted EUA for the Pfizer vaccine. Alarmingly, it was just days before the MHRA granted authorization in the UK on December 2, 2020. Veronika’s assumption that the ‘visible particles’ could be LNPs (lipid nanoparticles) is hard to accept given nanoparticles are not visible to the naked eye. Other anomalies were apparent, yet this was probably still a historical effort in terms of speed of vaccine development. It seems clear however some more time was needed.
How % mRNA integrity was apparently resolved
The discrepancy between batches appears to have may been resolved when it’s mentioned that the ‘latest lots [received by the FDA] indicate the % intact RNA are back around 70-75%.’
However, in a leaked report of a meeting with Pfizer and the EMA on November 26, 2020, a day after Veronika’s email, it shockingly reveals that the RNA integrity specification was revised down to >=50% for drug product shelf life, significantly lower than the minimum threshold of 70% that Acuitas Therapeutics had stipulated and the average 78% of the clinical batches. Was this the EMA’s (and potentially FDA/MHRA/HC) way of ‘resolving’ the issue to ensure ‘an approval by the end of the year’?

Mention is made of ‘uncertainties about consistency of product quality and hence uncertainty as regards product safety and efficacy of the commercial product.’ Yet, it’s baffling how lowering the RNA integrity specification would remedy that major objection.
In another slide the artifact states, ‘Truncated [shortened] and modified RNA species should be regarded as product- related impurities.’ This confirms that these shortened mRNA species which lowered the level of %mRNA integrity were classed as impurities. Another alarming concern arising from these impurities is flagged ‘the possibility of translated proteins other than intended spike protein (S1 S2) resulted from truncated and/or modified mRNA species should be addressed.’ (See screenshot below)

The evidence in this report confirms that regulatory bodies like the FDA, MHRA, EMA and Health Canada knew of the differences in batches, regarding % mRNA integrity and because of that the effect on ‘safety and efficacy’ was unknown. The leaked Pfizer/EMA meeting report raises material concerns assuming the issue was resolved by simply lowering the RNA integrity specification. In other words, perhaps it was never resolved.
A particular website that has drawn a lot of attention recently, which speaks to the difference between batches is howbadismybatch.com. It’s a comprehensive database with analysis on ‘batch codes and associated deaths, disabilities and illnesses for Covid 19 Vaccines.’ By entering a batch number of any of the Covid-19 vaccines, it tells you the frequency of adverse events reported associated with that batch.
I spoke with Sasha Latypova, who has run clinical trials for over 25 years and owns her own biotech company, to ask her expert opinion on the leaked documents. She said:
“The lack of mRNA integrity and presence of uncharacterized fragments of RNA in batches of Pfizer’s product was identified as a “Major Objection” – a formal regulatory red flag, deemed a product impurity and would have been a showstopper in any normal drug approval process. At a minimum, it required an additional “bridging” clinical trial to evaluate the clinical effects which would have taken months to design and conduct properly. Panic overruled scientific integrity, and an arbitrarily lowered batch acceptance standard was adopted for the sake of meeting a politically motivated deadline. To date, this issue remains unresolved and could be the underlying cause for the enormous variation in the rates of adverse events and deaths observed for different manufacturing batch numbers in the CDC VAERS and other databases.“
Latypova made an apt reference to the fate of the Titanic, by drawing a comparison in the way regulatory bodies conducted their ‘warp speed’ process of authorising the Covid-19 vaccines. The Titanic’s captain, Edward J. Smith, was aiming to better the crossing time of another vessel, which meant the ship was travelling way too fast, in waters known to have ice. This set it on a fatal collision with an iceberg and the rest is history.
In light of the evidence included in this report and the fact that the Pfizer-BioNTech Covid-19 vaccine is one of the most lucrative products in history (last year Pfizer made $37 billion in sales with predictions for 2022 being $32 billion), this author strives to open a discussion with some vital questions which must be addressed by the regulatory agencies involved, Pfizer and those in the scientific/medical community:
What are the safety and efficacy implications of a significantly lowered mRNA integrity (arising from truncated and modified mRNA) in the commercial batches of this vaccine?
Exactly what are the visible particles observed in the DP (drug product) that Pfizer shared last minute with the EMA, FDA and MHRA and what are its safety and efficacy implications?
Answers to these questions are of major public importance.
Originally published on Trial Site News.
Please see also: My leaked EMA emails report presentation to Dr Reiner Fuellmich & his Corona Investigative Committee on Sonia Elijah Substack here.