

Una investigación adicional sobre los correos electrónicos filtrados de EMA y los documentos confidenciales relacionados con la vacuna COVID-19 de Pfizer-BioNTech
En junio, Trial Site News publicó un informe de investigación explosivo sobre los correos electrónicos filtrados de la Agencia Europea de Medicamentos (EMA) y otros informes confidenciales relacionados con Pfizer, que expusieron hechos preocupantes en el período previo a la autorización de la vacuna Pfizer-BioNTech COVID-19. . Reveló:
- Una carrera impulsada políticamente entre reguladores clave en su prisa por autorizar la vacuna.
- A fines de noviembre de 2020, los reguladores, incluidos la FDA de EE. UU., la Agencia Europea de Medicamentos, Health Canada y la MHRA del Reino Unido, estaban al tanto de la pérdida significativa de la integridad del ARN de los lotes comerciales (~55% de integridad del ARNm) de la vacuna Pfizer-BioNTech en comparación a los clínicos (~78% de integridad del ARNm). Esto fue clasificado por la EMA como una «objeción importante» junto con las partículas visibles observadas, que se clasificaron como «impurezas».
- Una presentación de PowerPoint filtrada el 26 de noviembre de una reunión entre Pfizer-BioNTech y la EMA reveló cómo esta importante objeción se ‘resolvió’ sorprendentemente: la especificación de integridad del ARN simplemente se redujo al 50%, por lo tanto, se permitió la mitad de todas las moléculas de ARNm en los lotes comerciales. ser truncado (no intacto).
- Se desconocían las implicaciones potenciales de la pérdida de integridad del ARN en términos de seguridad y eficacia.
Este informe se centra en más correos electrónicos filtrados con una referencia específica a la comisaria europea, Úrsula von der Leyen, y el grado inusual en que estaba dispuesta a llegar para acorralar a los estados miembros (EM) para evitar el uso del artículo 5 (2) (su ley nacional de emergencia). Use la Autorización para las vacunas COVID-19, pero vaya con una Autorización de comercialización condicional (CMA) de la UE. Brinda luz sobre otros documentos confidenciales de EMA filtrados: la presentación de observaciones del CMC de la Oficina de calidad del 24 de noviembre de 2020 por parte del BWP (grupo de trabajo sobre productos biológicos) y el informe de evaluación de la revisión continua del relator, que revela más evidencia que respalda las «objeciones principales» discutidas en el informe original de Trial Site News. Examina las versiones no redactadas de los contratos de vacunas de Pfizer y el Acuerdo de compra anticipada de la CE firmado en noviembre de 2020 con Pfizer y BioNTech «para el desarrollo, la producción, las opciones de compra prioritarias y el suministro de una vacuna COVID-19 exitosa para los Estados miembros de la UE».
No es ajena al escándalo
Úrsula von der Leyen ha estado bajo un intenso escrutinio público sobre sus negociaciones privadas de un acuerdo de vacunas de miles de millones de euros (el contrato más grande de la UE) a través de mensajes de texto secretos y llamadas telefónicas realizadas con el director ejecutivo de Pfizer, Albert Bourla, contraviniendo la decisión de la Comisión de que un consejo de dirección ‘brinde orientación a lo largo del proceso de evaluación’. Además, se negó a otorgar acceso público a los mensajes de texto secretos intercambiados entre Bourla y ella, cuando se le pidió que lo hiciera. El Tribunal de Cuentas Europeo publicó un informe alarmante, que decía ‘solicitamos a la Comisión que nos proporcione información sobre las negociaciones preliminares de este acuerdo (expertos científicos consultados y asesoramiento recibido, calendario de las conversaciones, actas de las discusiones y detalles de los términos y condiciones acordados). Sin embargo, no llegó ninguno.
El fuego también se ha estado calentando para Bourla de Pfizer, cuando fue llamado a testificar ante el Comité Especial sobre COVID-19 del Parlamento Europeo para enfrentar preguntas sobre esos acuerdos secretos de vacunas, pero en el último minuto se retiró de enfrentar al comité.
Los correos filtrados
El informe de investigación original de Trial Site News destacó la enorme presión ejercida por la CE (Comisión Europea) sobre la EMA para otorgar CMA en un plazo muy acelerado. Se publicó un correo electrónico de Noel Wathion (exdirector ejecutivo adjunto de EMA), que reveló una TC (teleconferencia telefónica) ‘ bastante tensa’ con la comisaria europea (Úrsula von der Leyen) que fue ‘ a veces incluso un poco desagradable’ .
«Un retraso de varias semanas… no era fácilmente aceptable para la CE [Comisión Europea]», afirma Wathion. También revela ‘cómo las consecuencias políticas parecen ser demasiado altas incluso si el nivel «técnico» en los EM pudieran defender tal «retraso» para que el resultado de la revisión científica sea lo más sólido posible’.
A continuación se muestra un correo electrónico de Hilde Boone de la EMA, en el que se indica que » ella [von der Leyen] estará preparada para llamar personalmente a los ministros de salud pertinentes para evitar el uso del artículo 5 (2)».


Artículo 5 (2) vs CMA
El artículo 5, apartado 2, de la Directiva 2001/83 establece que “los Estados miembros podrán autorizar temporalmente la distribución de un medicamento no autorizado en respuesta a la propagación presunta o confirmada de agentes patógenos, toxinas, agentes químicos o radiación nuclear, cualquiera de los cuales podría causar daño.‘ En otras palabras, es el equivalente a una Autorización de Uso de Emergencia a nivel de estado miembro. Se puede hacer muy rápidamente porque el medicamento no necesita pasar por el proceso estándar de autorización nacional.
Un experto en derecho de la UE explicó a Trial Site News la desventaja de que los MS utilizasen el art. 5 (2), y es que habría dado lugar a una competencia entre los estados miembros, lo que daría lugar a un acceso/distribución desigual de las vacunas contra la COVID-19, en particular para aquellos Estados miembros que prefirieran esperar a la CMA de la UE (que lleva más tiempo ya que es se supone que debe seguir un marco controlado y sólido que proporciona salvaguardas). Una CMA de la UE ayudó a garantizar que las vacunas estuvieran disponibles para todos los estados miembros, al mismo tiempo, que era uno de los objetivos de la estrategia COVID-19 de la UE.
Sin embargo, ¿Podría haber otras razones por las que von der Leyen estaba desesperada por que los Estados miembros evitaran el Artículo 5 (2) para que ella estuviera dispuesta a llamar a todos los ministros de salud relevantes?
En el siguiente correo electrónico de Noel Wathion, afirma ‘ ya que la 1ª opción sigue siendo aspirar a una CMA en lugar de un Art 5 (2) junto o no a un Art 5 (3)…….’


El artículo 5, apartado 3, establece: «Los Estados miembros establecerán disposiciones para garantizar que los titulares de las autorizaciones de comercialización, los fabricantes y los profesionales sanitarios no estén sujetos a responsabilidad civil o administrativa por las consecuencias derivadas del uso de un medicamento…».
El hecho de que Wathion señale que un art. 5 (2) puede combinarse o no con un art. 5 (3), plantea la importante cuestión de si los estados miembros habrían tenido la opción de no indemnizar a los titulares de las autorizaciones de comercialización (en este caso, BioNTech) y a los fabricantes (BioNTech y Pfizer).
Los contratos predatorios de Pfizer y los acuerdos de compra anticipada
Dado lo que sabemos sobre los contratos de Pfizer filtrados y puestos a disposición por la organización sin fines de lucro de defensa del consumidor, Public Citizen; esta compañía farmacéutica aparentemente ha intimidado a los países para que guarden silencio; puede ir tras los bienes del estado soberano (bajo la renuncia a la inmunidad soberana) y disfrutar de una indemnización completa, exención de responsabilidad legal que pueda resultar de su producto, de hecho, es el Comprador quien se hace responsable en su nombre. Esto significa que los gobiernos han tenido que pagar una compensación a los ciudadanos que sufrieron un evento adverso de la vacuna, no al fabricante de la vacuna.
La siguiente captura de pantalla se tomó de la versión no redactada del acuerdo de compra anticipada (APA) entre la Comisión Europea (CE), actuando en representación y en nombre de los estados miembros, Pfizer Inc. y BioNTech (colectivamente ‘el Contratista’) firmado en Noviembre de 2020 (casi al mismo tiempo que se generaron los correos electrónicos de EMA filtrados). Establece que «cada Estado miembro participante indemnizará y eximirá de responsabilidad al Contratista, a sus Afiliados … de todas y cada una de las responsabilidades incurridas… en relación con los daños, perjuicios y pérdidas… que surjan o se relacionen con el uso y el despliegue de las Vacunas…”


Sin embargo, la información publicada en el sitio web de la CE establece que bajo una Autorización de comercialización condicional (CMA) de la UE, ‘la responsabilidad es del titular de la autorización de comercialización’ (que en este caso es BioNTech). Esto entra directamente en conflicto con el APA que la CE firmó con Pfizer y BioNTech (por 200 millones de dosis de vacuna a un precio de 15,50 euros por dosis sin IVA), un mes antes de que se concediera la CMA. Cabe señalar que la CE publicó una versión muy redactada de la misma APA, cualquier sección relacionada con indemnización/responsabilidad o simplemente considerada «sensible» ha sido censurada.
Otro punto importante a considerar cuando se trata del Art. 5 (2) frente a una CMA de la UE, es que dado que algunos países europeos exigieron la vacuna para adultos, grupos de edad en riesgo y ciertos sectores laborales, es muy poco probable que esos EM hubieran podido hacerlo con un medicamento no autorizado, como estas vacunas habrían sido clasificados en el Art 5 (2).
Los problemas de CMC
El correo electrónico de Wathion del 20 de noviembre de 2020 plantea varios puntos preocupantes: todavía hay problemas con ambos (CMC parece ser una preocupación para Pfizer/BioNTech y la revisión continua de Moderna acaba de comenzar dando menos tiempo para revisión), por lo que debe verse si todo esto se puede resolver a tiempo, sin comprometer la solidez de la revisión.‘
El informe original de Trial Site News discutió cuáles eran esos problemas de CMC (Chemistry Manufacturing and Controls) con Pfizer/BioNTech, particularmente la pérdida de integridad del ARN en los lotes comerciales y las partículas visibles desconocidas observadas. Wathion afirma de manera reveladora que ‘ todavía hay problemas’ habla de la noción de que estos ‘problemas’ no se resolvieron sino que habían estado en curso. Se enfatiza la preocupación de Wathion de ‘comprometer la solidez de la revisión’ debido a la necesidad de autorizar ‘a tiempo’. Esta preocupación de la velocidad por encima de la seguridad se refleja en otros correos electrónicos filtrados, particularmente de Wathion.
El informe también contenía un correo electrónico filtrado de Veronika Jekerle, Jefa de la Oficina de Calidad Farmacéutica de EMA, donde describió las 3 principales objeciones acordadas y la conclusión del BWP (Biologics Working Party) con respecto a la vacuna Pfizer-BioNTech. Su correo electrónico fue enviado el 24 de noviembre, el mismo día de la presentación de BWP. A continuación, hay una serie de capturas de pantalla de la presentación en power point de BWP filtrada (a la que Jekerle hace referencia en su correo electrónico), titulado «Observaciones CMC de la Oficina de calidad de EMA». Al final de su correo electrónico, agradece, ‘Ton, Brian y Claudio’. Ton van der Stappen y Brian Dooley se nombran en la captura de pantalla de la diapositiva a continuación.

La siguiente captura de pantalla presenta datos de gran alcance, que respaldan una de las principales objeciones de la EMA en referencia a la caída significativa en el porcentaje de integridad del ARN entre los lotes clínicos y comerciales. Cabe señalar que se informó que los lotes de los sitios de Pfizer en Andover, EE. UU. (~62 %) y Puurs, Bélgica (~55 %) tenían un porcentaje de integridad de ARN significativamente más bajo que los lotes proporcionados por BNT (BioNTech) y Polymun.
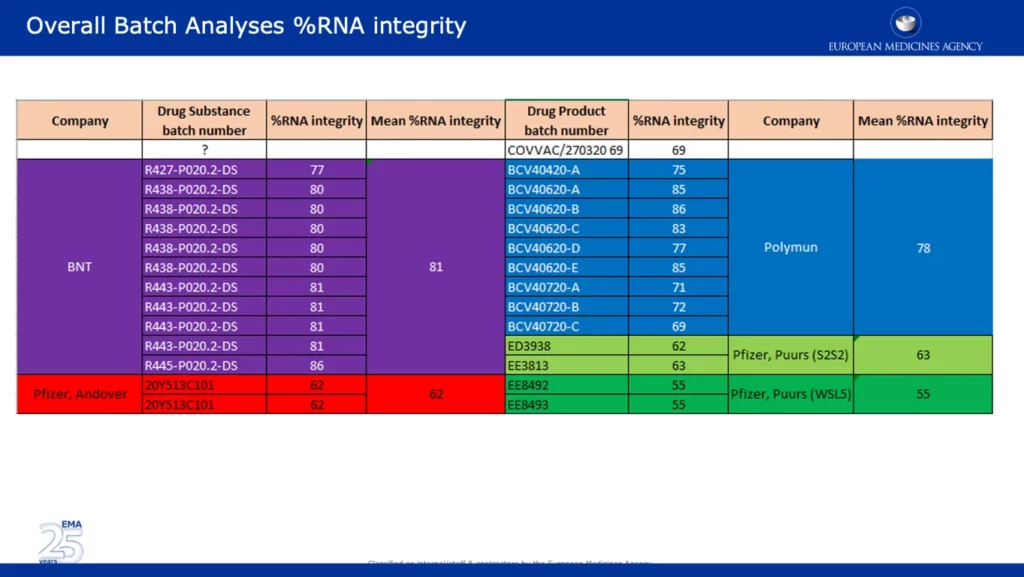
La captura de pantalla a continuación se tomó del APA no redactado firmado por la CE. Especifica que la mayor parte del suministro de vacunas de Europa provendrá del sitio de fabricación de Pfizer en Puurs, Bélgica. Esto es preocupante dado que los datos muestran que el porcentaje de integridad de ARN de los lotes de ese sitio fue el más bajo con un 55 %, en comparación con otros sitios.


La siguiente captura de pantalla muestra los criterios de aceptación de varios lotes de producto farmacéutico BNT 162B2. Producto farmacéutico se refiere a “medicamento en su forma comercializada, incluidos sus rellenos, agentes colorantes y otros agentes activos o inactivos ‘. El criterio de aceptación para el suministro de emergencia es mayor o igual al 50 %, que está justo por debajo de los lotes con el porcentaje de integridad de ARN más bajo suministrados por Pfizer, Puurs. Por alguna razón, los criterios de aceptación para los lotes de productos farmacéuticos clínicos difieren y se establecen más altos en un 60 % o más.

CMC (Química, Fabricación y Controles) ‘ involucra la definición de prácticas de fabricación y especificaciones del producto que deben seguirse y cumplirse para garantizar la seguridad del producto y la consistencia entre lotes. Dados los datos que se muestran arriba, es evidente que hubo problemas significativos de CMC con respecto a la inconsistencia del porcentaje de integridad de ARN entre lotes, lo que se refleja en los correos electrónicos y documentos filtrados de la EMA. Lo preocupante es que el fabricante (Pfizer/BioNTech) afirmó: «La eficacia del medicamento depende de la expresión del ARN administrado, que requiere una molécula de ARN suficientemente intacta».
Es desconcertante cómo la reducción de la especificación al 50% fue potencialmente la forma en que se ‘resolvió’ esta importante objeción. Según una evaluación de revisión continua del relator filtrada (fecha del informe revisado: 25 de noviembre de 2020) compilada por los relatores del CHMP y el PRAC, encontraron que los «criterios de aceptación actuales» son particularmente preocupantes. El informe establece que «no se presentan datos de caracterización sobre la integridad del ARN y las formas truncadas y no se abordan los posibles riesgos de seguridad asociados con las isoformas de ARN truncado«. Esto es especialmente importante teniendo en cuenta que los criterios de aceptación actuales de DS y DP permiten hasta un 50 % de especies fragmentadas.’


El ARN truncado (acortado por la falta de la sección superior o final) se define como una «impureza relacionada con el producto» y el hecho de que los posibles riesgos de seguridad que surgen de esta especie fragmentada «no se aborden» es muy alarmante.
Se hace referencia a ‘los criterios de aceptación actuales de DS y DP’, esto implica que no siempre se fijó en ese nivel y tal vez se había cambiado (rebajado). La pregunta es ¿por qué? ¿Podría haber sido que el proceso 2 (la fabricación de los lotes comerciales) simplemente no era replicable al mismo nivel de especificación a los lotes clínicos (pequeña escala) del proceso 1, por lo tanto, se estableció un estándar más bajo para que se otorgara la CMA?
Trial Site News se comunicó con la EMA con respecto al contenido de los correos electrónicos y documentos filtrados. La pronta respuesta de la oficina de prensa de la EMA se publica a continuación en su totalidad.
“La investigación del material publicado reveló que la correspondencia fue manipulada por los perpetradores antes de la publicación. No todos los documentos fueron publicados en su forma original integra y pueden haber sido sacados de contexto. Si bien los correos electrónicos individuales eran auténticos, se seleccionaron y agregaron datos de diferentes usuarios, se crearon capturas de pantalla de varias carpetas y buzones de correo y los perpetradores agregaron títulos adicionales.
Estos documentos no presentan una imagen completa de la evaluación de Comirnaty, la vacuna COVID-19 desarrollada por BioNTech/Pfizer. Muestran la situación hasta principios de diciembre de 2020, cuando se descubrió el hackeo, pero no mencionan la cantidad considerable de datos, información y aclaraciones adicionales presentados por BioNTech/Pfizer hasta el 21 de diciembre de 2020, día en que el comité de medicamentos de uso humano (CHMP) de la EMA dio su recomendación de conceder una autorización de comercialización para esta vacuna.
Comirnaty funciona porque el ARNm que contiene proporciona instrucciones para producir una proteína de pico que desencadena una respuesta inmunitaria. Por tanto, su eficacia depende de la presencia de una cantidad adecuada de ARNm intacto, que se sabe que es relativamente inestable. Lo que muestran los documentos es cómo funciona la evaluación de cualquier medicamento: tras el examen de los datos presentados por la empresa, el CHMP tenía dudas sobre la integridad del ARNm y las planteó formalmente como una «objeción importante». Esta es una parte integral de la evaluación de cualquier medicamento. Si quedan objeciones importantes sin resolver, impedirían la concesión de la autorización de comercialización. En este caso, la empresa atendió satisfactoriamente las cuestiones planteadas y posteriormente suministró la información y datos requeridos con posterioridad a principios de diciembre de 2020.
El informe de evaluación pública de Comirnaty resume las conclusiones del CHMP sobre este tema y detalla los pasos tomados durante el procedimiento de autorización de comercialización de Comirnaty, así como las obligaciones impuestas al titular de la autorización de comercialización de realizar estudios adicionales para monitorear de cerca la calidad farmacéutica de la vacuna. Estas obligaciones también se incluyeron en la información del producto publicada en el momento de la opinión del CHMP.
Incluso en una emergencia de salud pública como la COVID-19, siempre ha habido consenso en toda la UE para no comprometer los estándares y basar cualquier recomendación en la evidencia científica disponible sobre la seguridad, la calidad farmacéutica y la eficacia de una vacuna, y nada más. Las autorizaciones solo se conceden cuando la evidencia muestra de manera convincente que los beneficios de la vacunación son mayores que los riesgos que plantea una vacuna”.
Publicado originalmente en Trial Site News
Sugerir una corrección




