

Un’ulteriore indagine sulle e-mail EMA trapelate e sui documenti riservati del vaccino Pfizer-BioNTech COVID-19
Di Sonia Elijah
A giugno, Trial Site News ha pubblicato un rapporto investigativo bomba sulle e-mail trapelate dell’Agenzia europea per i medicinali (EMA) e altri rapporti riservati relativi a Pfizer, che hanno esposto fatti relativi alla corsa all’autorizzazione del vaccino Pfizer-BioNTech COVID-19. Il report ha rivelato:
- Una corsa guidata politicamente tra le autorità di regolamentazione chiave nella loro corsa ad autorizzare il vaccino.
- Entro la fine di novembre 2020, le autorità di regolamentazione, tra cui l’FDA statunitense, l’Agenzia europea per i medicinali, Health Canada e MHRA del Regno Unito, erano tutte consapevoli della significativa perdita di integrità dell’RNA dei lotti commerciali (~55% di integrità dell’mRNA) del vaccino Pfizer-BioNTech rispetto a quelli clinici (~78% di integrità dell’mRNA). Questa è stata classificata dall’EMA come una “grande obiezione” insieme alle particelle visibili osservate, che sono state classificate come “impurità”.
- Una presentazione PowerPoint trapelata il 26 novembre di un incontro tra Pfizer-BioNTech e l’EMA ha rivelato come questa importante obiezione sia stata “risolta” in modo scioccante: la specifica di integrità dell’RNA è stata semplicemente ridotta al 50%, quindi era consentito che la metà di tutte le molecole di mRNA nei lotti commerciali fosse troncata (non integra).
- Le potenziali implicazioni della perdita di integrità dell’RNA in termini di sicurezza ed efficacia erano sconosciute.
Questo rapporto si concentra su ulteriori e-mail trapelate con specifico riferimento al Commissario europeo, Ursula von der Leyen e l’insolita misura in cui la Von Der Leyen era disposta a rinchiudere gli Stati membri (SM) per evitare l’uso dell’articolo 5 (2) (autorizzazione per uso d’emergenza per i vaccini COVID-19), procedendo piuttosto con un’autorizzazione all’immissione in commercio condizionale dell’UE (CMA).
Questo getta luce su altri documenti sensibili dell’EMA trapelati: la presentazione delle osservazioni del CMC dell’Ufficio per la qualità del 24 novembre 2020 da parte del BWP (gruppo di lavoro sui biologici) e il rapporto di valutazione del Rolling Review del relatore, che rivelava ulteriori prove a sostegno delle “obiezioni principali” discusse nel report originale di Trial Site News. Esamina le versioni non modificate dei contratti sui vaccini Pfizer e l’accordo di acquisto anticipato della CE firmato nel novembre 2020 con Pfizer e BioNTech “per lo sviluppo, la produzione, le opzioni di acquisto prioritario e la fornitura di un vaccino COVID-19 di successo per gli Stati membri dell’UE”.
Non estranea allo scandalo
Ursula von der Leyen è stata sottoposta a un intenso controllo pubblico sulle sue negoziazioni private di un accordo multimiliardario sui vaccini (il più grande contratto dell’UE) tramite messaggi segreti e telefonate fatte con l’amministratore delegato di Pfizer, Albert Bourla, contrariamente alla decisione della Commissione per un consiglio direttivo che potesse “fornire una guida durante tutto il processo di valutazione”. Una Corte dei conti europea ha pubblicato un rapporto allarmante, in cui si afferma che “abbiamo chiesto alla Commissione di fornirci informazioni sui negoziati preliminari per questo accordo (esperti scientifici consultati e pareri ricevuti, tempi dei colloqui, resoconti delle discussioni e dettagli i termini e le condizioni concordati). Tuttavia, nulla è arrivato.’
Il fuoco si è acceso anche per Bourla della Pfizer, quando è stato chiamato a testimoniare davanti alla commissione speciale del Parlamento europeo sul COVID-19 per affrontare le domande su quegli accordi segreti sui vaccini, ma all’ultimo minuto ha rifiutato di affrontare la commissione.
Le email trapelate
Il rapporto investigativo originale di Trial Site News ha evidenziato l’enorme pressione esercitata dalla CE (Commissione Europea) sull’EMA per concedere CMA (Autorizzazione all’immissione in commercio condizionale) con tempistiche molto accelerate. È stata pubblicata un’e-mail di Noel Wathion (ex vicedirettore esecutivo dell’EMA), che ha rivelato una teleconferenza piuttosto tesa con il commissario europeo (Ursula von der Leyen) che è stata “a volte anche un po’ spiacevole”.
“Un ritardo di diverse settimane… non era facilmente accettabile per la CE [Commissione europea]”, afferma Wathion. Rivela anche “come le ricadute politiche sembrano essere troppo elevate anche se il livello “tecnico” degli Stati membri potrebbe difendere un tale “ritardo” al fine di rendere il risultato della revisione scientifica il più solido possibile”.
Di seguito è riportata un’e-mail di Hilde Boone dell’EMA, in cui si afferma che “lei [von der Leyen] sarà disposta a chiamare personalmente i ministri della salute competenti per evitare l’uso dell’articolo 5, paragrafo 2.”.

Articolo 5 (2) vs CMA
L’articolo 5, paragrafo 2, della direttiva 2001/83 recita: «Gli Stati membri possono autorizzare temporaneamente la distribuzione di un medicinale non autorizzato in risposta alla diffusione sospetta o confermata di agenti patogeni, tossine, agenti chimici o radiazioni nucleari che potrebbero causare danni”. In altre parole, è l’equivalente di un’autorizzazione all’uso di emergenza a livello di stato membro. Può essere fatto molto rapidamente perché il medicinale non ha bisogno di passare attraverso il processo di autorizzazione nazionale standard.
Un esperto di diritto dell’UE ha spiegato a Trial Site News lo svantaggio degli Stati membri ad utilizzare l’art. 5 (2); ciò avrebbe dato luogo a concorrenza tra gli Stati membri, portando a un accesso/distribuzione iniquo dei vaccini COVID-19, in particolare per quegli Stati membri che hanno preferito attendere l’autorizzazione all’immissione in commercio condizionale dell’UE (che richiede più tempo poiché dovrebbe seguire un quadro controllato e solido che fornisca garanzie). Una CMA dell’UE ha contribuito a garantire che i vaccini sarebbero stati disponibili per tutti gli Stati membri, allo stesso tempo, che era uno degli obiettivi della strategia dell’UE COVID-19.
Tuttavia, potrebbero esserci stati ulteriori motivi per cui von der Leyen desiderava disperatamente che gli Stati membri evitassero l’articolo 5 (2) al punto da essere disposta a chiamare lei stessa ogni ministro della salute competente?
Nell’e-mail di seguito di Noel Wathion, afferma “poiché la prima opzione è ancora quella di puntare a un CMA piuttosto che a un articolo 5 (2) accoppiato o meno a un articolo 5 (3)..’

L’articolo 5, paragrafo 3, recita: “Gli Stati membri stabiliscono disposizioni al fine di garantire che i titolari delle autorizzazioni all’immissione in commercio, i fabbricanti e gli operatori sanitari non siano soggetti a responsabilità civile o amministrativa per le conseguenze derivanti dall’uso di un medicinale…”.
Il fatto che Wathion rilevi che un art. 5, comma 2, possono essere abbinati o meno ad un art. 5 (3), solleva l’importante questione se gli Stati membri avrebbero avuto la possibilità di non dare indennizzo ai titolari delle autorizzazioni all’immissione in commercio (in questo caso BioNTech) e ai produttori (BioNTech e Pfizer).
I contratti Pfizer predatori e gli accordi di acquisto anticipato
Quello che sappiamo sui contratti Pfizer trapelati è stato messo a disposizione dall’organizzazione senza scopo di lucro per la difesa dei consumatori, Public Citizen; questa azienda farmaceutica ha apparentemente costretto i paesi al silenzio; può perseguire beni statali sovrani (con la rinuncia all’immunità sovrana) e godere di un indennizzo totale – esenzione dalla responsabilità legale che potrebbe derivare dal loro prodotto – infatti è l’Acquirente che è responsabile per loro conto. Ciò significa che i governi, e non i produttori dei vaccini, hanno dovuto pagare un risarcimento ai cittadini che hanno subito un evento avverso del vaccino.
Lo screenshot seguente è tratto dalla versione non redatta dell’accordo di acquisto anticipato (APA) tra la Commissione Europea (CE), che agisce per conto e in nome degli Stati membri, Pfizer Inc. e BioNTech (collettivamente “l’Appaltatore”) firmato Novembre 2020 (più o meno nello stesso periodo in cui sono state generate le e-mail EMA trapelate). Afferma che “ciascuno Stato membro partecipante indennizzerà e manleverà l’Appaltatore, le sue affiliate… da e contro qualsiasi responsabilità sostenuta… relativa a danni, danni e perdite… derivanti o relativi all’uso e alla diffusione dei vaccini ..’

Tuttavia, le informazioni pubblicate sul sito Web della CE affermano che ai sensi di un’autorizzazione all’immissione in commercio condizionale dell’UE (CMA), “la responsabilità è del titolare dell’autorizzazione all’immissione in commercio” (che in questo caso è BioNTech). Ciò è in conflitto diretto con l’APA che la CE ha firmato con Pfizer e BioNTech (per 200 milioni di dosi di vaccino al prezzo di 15,50 euro per dose IVA esclusa), un mese prima che fosse concesso il CMA. È interessante notare che la CE ha pubblicato una versione pesantemente redatta dell’identico APA, qualsiasi sezione relativa a indennizzo/responsabilità o semplicemente considerata “sensibile” è stata censurata.
Un altro punto importante da considerare quando si tratta dell’articolo 5, paragrafo 2 rispetto a una CMA dell’UE, è che, dato che alcuni paesi europei hanno imposto il vaccino per gli adulti, i gruppi di età a rischio e alcuni settori di lavoro, è altamente improbabile che gli Stati membri sarebbero stati in grado di farlo con un medicinale non autorizzato, e che questi vaccini sarebbero stati classificati ai sensi dell’articolo 5, paragrafo 2.
I problemi della CMC
L’e-mail di Wathion del 20 novembre 2020 solleva diversi punti preoccupanti: “ci sono ancora problemi con entrambi (CMC sembra essere una preoccupazione per Pfizer/BioNTech e la revisione continua per Moderna ha appena iniziato a concedere meno tempo per la revisione), quindi deve essere visto se tutto questo può essere risolto in tempo, senza compromettere la solidità della revisione.’
Il rapporto originale di Trial Site News discuteva quali fossero quei problemi CMC (Chimica, produzione e controllo) con Pfizer/BioNTech, in particolare la perdita di integrità dell’RNA nei lotti commerciali e le particelle visibili sconosciute osservate. Wathion afferma in modo significativo che “ci sono ancora problemi” parla dell’idea che questi “problemi” non fossero stati risolti, ma che fossero in corso. Viene sottolineata la preoccupazione di Wathion di “compromettere la solidità della revisione” a causa della necessità di autorizzare “in tempo”. Questa preoccupazione per la velocità rispetto alla sicurezza si riflette in altre e-mail trapelate, in particolare da Wathion.
Il rapporto conteneva anche un’e-mail trapelata da Veronika Jekerle, Head of Pharmaceutical Quality Office dell’EMA, in cui ha delineato le 3 principali obiezioni concordate e la conclusione del BWP (Biologics Working Party) in merito al vaccino Pfizer-BioNTech. La sua e-mail è stata inviata il 24 novembre, lo stesso giorno della presentazione del BWP. Di seguito, una serie di schermate dell’effettiva presentazione in power point BWP trapelata (a cui Jekerle fa riferimento nella sua e-mail), intitolata “Osservazioni CMC dell’ufficio qualità EMA”. Alla fine della sua e-mail ringrazia “Ton, Brian e Claudio”. Ton van der Stappen e Brian Dooley sono nominati nello screenshot della diapositiva qui sotto.

La schermata sottostante presenta dati di ampia portata, che confermano una delle principali obiezioni dell’EMA in riferimento al significativo calo della %RNA integrity tra i lotti clinici e quelli commerciali. È degno di nota il fatto che i lotti provenienti dai siti di Pfizer di Andover, USA (~62%) e Puurs, Belgio (~55%) sono stati segnalati come aventi una % di integrità dell’RNA significativamente inferiore rispetto ai lotti forniti da BNT (BioNTech) e Polymun.
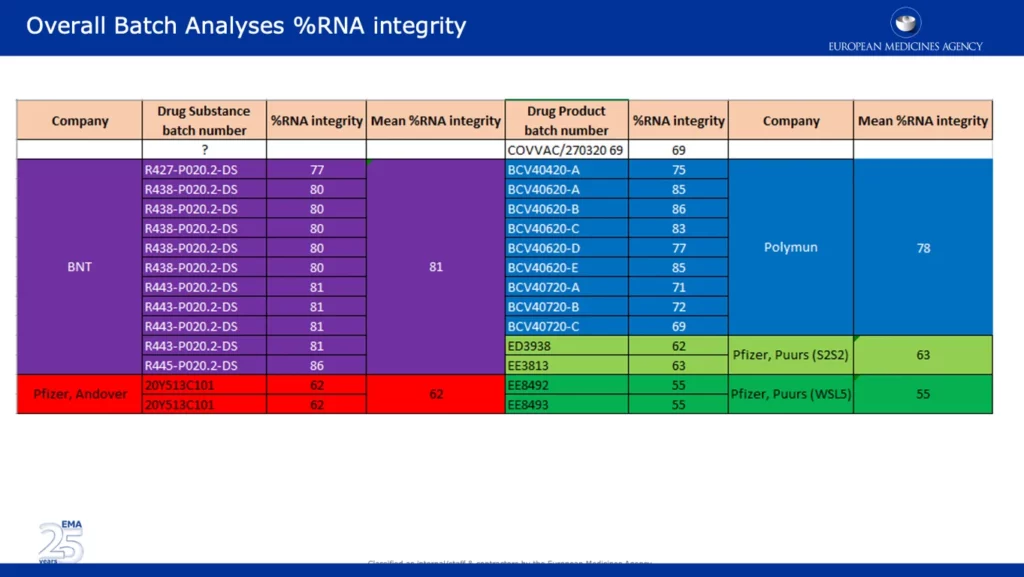
Lo screenshot qui sotto è tratto dall’APA non redatto firmato dalla CE. Specifica che la maggior parte della fornitura di vaccini in Europa proverrà “dal sito di produzione di Pfizer a Puurs, in Belgio”. Ciò è preoccupante dato che i dati mostrano che l’integrità dell’%RNA dei lotti di quel sito era il più basso al 55%, rispetto ad altri siti.

La schermata sottostante mostra i criteri di accettazione di vari lotti di prodotti farmaceutici BNT 162B2. Per prodotto farmaceutico si intende “il farmaco nella sua forma commercializzata, compresi i riempitivi, i coloranti e altri agenti attivi o inattivi”. Il criterio di accettazione per l’Emergency Supply è maggiore o uguale al 50%, che si trova appena al di sotto dei lotti con la più bassa integrità di %RNA forniti da Pfizer, Puurs. Per qualche motivo, i criteri di accettazione per i lotti di prodotti farmaceutici clinici sono diversi e sono più alti, maggiori o uguali al 60%.

CMC (Chimica, produzione e controlli) “comporta la definizione delle pratiche di produzione e delle specifiche del prodotto che devono essere seguite e soddisfatte per garantire la sicurezza del prodotto e la coerenza tra i lotti”. Dati i dati mostrati sopra, è evidente che c’erano problemi significativi di CMC riguardo all’incoerenza dell’integrità della percentuale di RNA tra i batch, che si riflette nelle e-mail e nei documenti EMA trapelati. La cosa preoccupante è che il produttore (Pfizer/BioNTech) ha affermato: “L’efficacia del prodotto farmaceutico dipende dall’espressione dell’RNA fornito, che richiede una molecola di RNA sufficientemente intatta”.
È sconcertante come l’abbassamento della specifica fino al 50% sia stato potenzialmente il modo in cui questa importante obiezione è stata “risolta”. ‘criteri di accettazione attuali’ particolarmente preoccupanti. Il rapporto afferma che “non vengono presentati dati di caratterizzazione sull’integrità dell’RNA e sulle forme troncate e non vengono affrontati i potenziali rischi per la sicurezza associati alle isoforme di RNA troncato“. Ciò è particolarmente importante considerando che gli attuali criteri di accettazione di DS e DP consentono fino al 50% di specie frammentate.‘

L’RNA troncato (accorciato dalla mancanza della sezione superiore o finale) è definito come “impurità relativa al prodotto” e il fatto che i potenziali rischi per la sicurezza derivanti da questa specie frammentata “non vengano affrontati” è altamente allarmante.
Si fa riferimento ai “criteri di accettazione DS e DP attuali“, ciò implica che non è stato sempre impostato quel livello e forse è stato modificato (abbassato). La domanda è perché? Potrebbe essere accaduto che il processo 2 (la produzione dei lotti commerciali) non fosse replicabile allo stesso livello di specifica dei lotti clinici (piccola scala) del processo 1, quindi è stato fissato uno standard inferiore per ottenere la CMA?
Trial Site News ha comunicato con l’EMA in merito al contenuto delle e-mail e dei documenti trapelati. Di seguito è stata pubblicata integralmente la pronta risposta dell’ufficio stampa dell’EMA.
“L’indagine sul materiale pubblicato ha rivelato che la corrispondenza è stata manipolata dagli autori prima della pubblicazione. Non tutti i documenti sono stati pubblicati nella loro forma originale e integrale e potrebbero essere stati presi fuori contesto. Sebbene le singole e-mail fossero autentiche, i dati di utenti diversi sono stati selezionati e aggregati, sono stati creati screenshot da più cartelle e caselle di posta e gli autori hanno aggiunto titoli aggiuntivi.
Questi documenti non presentano un quadro completo della valutazione di Comirnaty, il vaccino COVID-19 sviluppato da BioNTech/Pfizer. Mostrano la situazione fino all’inizio di dicembre 2020, quando l’hack è stato scoperto, ma non menzionano la notevole quantità di dati aggiuntivi, informazioni e chiarimenti presentati da BioNTech/Pfizer fino al 21 dicembre 2020, giorno in cui il comitato per l’uomo dell’EMA medicinali (CHMP) ha raccomandato di concedere un’autorizzazione all’immissione in commercio per questo vaccino.
Comirnaty funziona perché l’mRNA che contiene fornisce istruzioni per produrre una proteina spike che innesca una risposta immunitaria. La sua efficacia dipende quindi dalla presenza di un’adeguata quantità di mRNA intatto, noto per essere relativamente instabile. Ciò che mostrano i documenti è come funziona la valutazione di qualsiasi medicinale: in seguito all’esame dei dati presentati dall’azienda, il CHMP ha sollevato domande sull’integrità dell’mRNA e le ha sollevate formalmente come “importante obiezione”. Questa è parte integrante della valutazione di qualsiasi medicinale. Se le obiezioni importanti rimangono irrisolte, precludono il rilascio dell’autorizzazione all’immissione in commercio. In questo caso, l’azienda ha affrontato le questioni sollevate in modo soddisfacente e successivamente ha fornito le informazioni ei dati richiesti dopo l’inizio di dicembre 2020, il che ha consentito all’EMA di muoversi verso un parere positivo per questo vaccino.
Il rapporto di valutazione pubblica di Comirnaty riassume le conclusioni del CHMP su questo tema e descrive in dettaglio le misure adottate durante la procedura di autorizzazione all’immissione in commercio di Comirnaty, nonché gli obblighi imposti al titolare dell’autorizzazione all’immissione in commercio di condurre ulteriori studi per monitorare da vicino la qualità farmaceutica di vaccino. Tali obblighi sono stati inclusi anche nelle Informazioni sul prodotto pubblicate al momento del parere del CHMP.
Anche in un’emergenza sanitaria pubblica come il COVID-19, c’è sempre stato consenso in tutta l’UE a non compromettere gli standard e a basare qualsiasi raccomandazione sulle prove scientifiche disponibili sulla sicurezza, la qualità e l’efficacia farmaceutica di un vaccino e nient’altro. Le autorizzazioni vengono concesse solo quando le prove dimostrano in modo convincente che i benefici della vaccinazione sono maggiori di qualsiasi rischio rappresentato da un vaccino.
Originariamente pubblicato su Trial Site News.
Suggerisci una correzione




