

Brev från underjorden
Bild: Shutter Stock
Läckta e-postmeddelanden från EMA, uppdaterat
I slutet av 2020 läckte en samling dokument – cirka 900 sidor från Pfizers ”vaccin”-avsnittet Chemistry Manufacturing and Controls (CMC) i den regulatoriska ansökan till Europeiska läkemedelsmyndigheten (EMA) – och skickades till ett antal journalister. Dokumenten innehöll också e-posttrafik från några av granskarna och högre chefer vid EMA. Läckan rapporterades av British Medical Journal som kunde bekräfta att dokumenten var äkta. Jag fick dessa dokument av en kollega ungefär ett år senare, i slutet av 2021, och jag läste och använde många av dem i mina analyser. Jag har sett många FoU-relaterade dokument från Pfizer i mitt professionella arbete, så jag kan också bekräfta att dessa läckta dokument i hög grad överensstämmer med typisk Pfizer-dokumentation. EMA förnekade inte äktheten, utan uppgav endast att rubrikerna i vissa av e-postmeddelandena hade ändrats.
Jag har ett mycket större antal följare idag och en mycket större förståelse för organisationen och den pseudo-juridiska strukturen hos den kriminella kartell som driver den globala grymhet som i vardagligt tal kallas ”covid pandemiresponsen”. Jag återkommer till de läckta e-postmeddelandena eftersom jag anser att de ger några mycket viktiga bevis.
EMA:s e-postfiler som jag har läst innehåller 14 skärmdumpar av e-postmeddelanden från mitten till slutet av november 2020. Mailen är från EMA:s personal och högre chefer. Enligt min åsikt visar dessa e-mails att:
- EMA:s granskare stod under ett massivt politiskt tryck för att hitta på nya sätt att godkänna de farliga produkter som inte kunde godkännas. Trycket utgick från de allra högsta nivåerna i USA:s, storbrittaniens och EU ländernas regeringar.
- EU:s kommissionär Ursula von der Leyen gav löften till medlemsstaterna som hon aldrig hade för avsikt att uppfylla för att binda dem alla till en enda pakt för vaccinavtal och på så sätt förhindra alla oberoende beslut i de enskilda länderna.
- Det fanns allvarliga och olösliga problem – med tanke på den avsiktligt orealistiska tidsplanen – med kvaliteten på den produkt som EMA:s personal pressades att godkänna. Vissa var obekväma med att göra detta och uttryckte sina farhågor. Andra ”förbisåg” tydligt påhittade uppgifter.
I slutändan spelade själva granskningen av lagstiftningen och de problem som togs upp ingen roll – produkten skulle marknadsföras oavsett vad som hände. Vi vet nu exakt varför – tillsynsmyndigheterna hade inte den föreskrivande makten över produkten. Läkemedelstillsynsmyndigheterna övervakar inte militärt material som kallas ”motåtgärder” och ”tillverkningsdemonstrationer” (ett lurigt uttryck för biologiska stridsmedel som tillverkas av den korrupta amerikanska regeringen och dess globala partners). E-mailen visar att majoriteten av EMA:s personal var omedvetna aktörer i denna pjäs.
Detta bekräftades nyligen vad gäller Storbritannien.
Baserat på svaret på MHRA FOIA:
”Alla Covid-vaccin- och terapeutiska godkännandebeslut fattades av licensministern och delegerades inte.”
Med andra ord – normalt sett delegeras befogenhet att granska och godkänna nya läkemedel formellt till MHRA från hälsoministern (Storbritannien). När det gäller covidprodukter uteblev denna delegering av befogenhet. Det verkar som om alla dessa åtgärder har satts in på egen hand av Matt Hancock (även om han pekar finger åt någon högre upp). Samma sak hände i USA – Alex Azar under Trumpadministrationen lät sprida detta farliga biomaterial bland amerikaner, och Xavier Becerra under Biden fortsätter att göra så i dag.
Politisk påtryckning.
Följande e-postväxling ägde rum den 16 november 2020 mellan högre chefer vid EMA:
Noel Wathion – biträdande verkställande direktör (pensionerad i juni 2021):

Agnes Saint-Raymond – chef för avdelningen för internationella frågor:

Marco Cavaleri, ordförande för arbetsgruppen för Covid-19-pandemier vid EMA:

E-postmeddelandena bör läsas nerifrån och upp.

Ett par intressanta saker: De tre tillsynsmyndigheterna – amerikanska FDA, brittiska MHRA och EU:s EMA – är alla upptagna med att samordna tidpunkten för godkännande innan någon formell granskning av data har ägt rum, innan rådgivande kommittéer har sett resultaten av de kliniska prövningarna, diskuterat dem, röstat om dem osv. De diskuterar tidpunkten för godkännande eftersom data inte spelar någon roll för om dessa produkter ska släppas ut på marknaden eller inte. Dessutom interagerar de som om de inte är tre separata organ för separata suveräna nationer, som är ansvariga för separata grupper av skattebetalare och med skilda kongress/parlamentarisk tillsyn, utan istället helt enkelt som byråkratiska avdelningar som redan har slagits samman till en global regering. Slutligen kom FDA att ”skynda sig in i EUA”, eftersom de ”pressas av Azar” (Alex Azar – HHS-sekreterare vid den tidpunkten) och ”Trump drar i trådarna”.
Många frågar mig hur det är möjligt att tusentals människor deltog i den bluff som iscensattes som ”covid pandemic response” – det är väl inte möjligt att så många människor är i maskopi med varandra! Men det var aldrig nödvändigtvis så många som faktiskt visste. Här är Noel Wathion, en av EMA:s högsta chefer, antingen inte medveten om att granskningen av data är irrelevant för om injektionerna kommer att släppas ut på marknaden, eller så ger han skickligt en felaktig bild av detta (jag tror faktiskt att han inte visste). Därför behöver inte heller EMA-personal under honom vara medvetna om detta, utan har helt enkelt skyndat sig för att utföra uppgiften de tilldelats. Kompartmentalisering är nyckeln till att dölja alla större bedrägerier inom stora organisationer och komplexa strukturer. Är detta skälet till att Noel Wathion avgick/pensionerade sig kort efter att de dödliga injektionerna lanserades? Han pressades också av EU-kommissionen att godkänna. Och Pfizer vill nu ha ett fullständigt marknadsföringstillstånd (MA) i stället för ett villkorligt tillstånd (CMA)! Observera – CMA utfärdades, men villkoren uppfylldes aldrig av Pfizer/BioNTech, för vem bryr sig, det var ett spel från början.
”(Co)-Rapps” = projektsekreterare. EMA är en europeisk enhet som består av medlemsstaternas tidigare separata ”behöriga myndigheter” som tidigare brukade reglera och godkänna läkemedel i varje land separat. I den europeiska strukturen väljs en teknisk granskningsgrupp och medgranskningsgrupp ut för en specifik produkt. I fallet med covid-”vacciner” var den svenska gruppen under ledning av Philip Josephson föredragande (huvudgranskare) och den franska gruppen under ledning av Jean-Michel Race projektsekreterare. ”CHMP” = Kommittén för humanläkemedel (vid EMA).

E-postmeddelandet är adresserat till Olga Solomon vid EU-kommissionen, och Noels chef, Emer Cooke, verkställande direktör för EMA och tidigare hög chef vid WHO, har fått en kopia. Här är Emer Cooke:

Ursulas smarta pakt.
Minns du henne?

Ursula von der Leyen – EU-kommissionär, som bland annat har förhandlat fram otroligt katastrofala leveransavtal med Pfizer för alla EU:s medlemsstaters räkning via sms med Pfizers vd Albert Bourla. I dessa avtal var EU-länderna tvungna att ställa statliga tillgångar som säkerhet, avstå från all kvalitetskontroll, import och konsumentskyddslagstiftning och ge upp sin nationella suveränitet – dvs. de fick inte ändra lagstiftningen om vaccinansvar i sina egna parlament! De rövarkontrakt som var komplett censurerade för att skydda så kallade ”Pfizers kommersiella intressen”. Följande e-postväxling har att göra med Ursulas tappra ansträngningar:

Det används en mängd akronymer, men de mest relevanta är ”EC” = EU-kommissionen, ”MS” = medlemsstaterna, ”EP” = Europaparlamentet. Den viktigaste meningen är att Ursula är ”beredd att personligen ringa upp berörda hälsoministrar för att undvika att artikel 5.2 används” . Vad handlar detta om? Artikel 5.2 hänvisar till ”artikel 5.2 i direktiv 2001/83” – tillstånd för användning vid nödsituationer i en europeisk medlemsstat, som ges av varje medlemsstat separat i sitt eget land. CMA är ett villkorligt marknadstillstånd som utfärdas av EMA för alla EU-medlemmar samtidigt. Förespråkas av EU som en mycket mer robust process än ett EUA (betoning tillagd av mig):
…CMA följer ett kontrollerat och robust ramverk som ger skyddsåtgärder som tillstånd för användning i nödsituationer inte ger. I själva verket är ett tillstånd för användning i nödsituationer inte ett godkännande av vaccinet utan ett godkännande av tillfällig användning av det otillåtna vaccinet. CMA säkerställer att all säkerhetsövervakning, tillverkningskontroller, inklusive kontroller av vaccinbatcher och andra skyldigheter efter godkännande tillämpas på ett rättsligt bindande sätt […]. Särskilt:
-Insatsen säkerställer en rigorös övervakning, genom EU:s system för säkerhetsövervakning, av läkemedlets säkerhet i hela EU. […]
– CMA säkerställer att produktens säkerhet övervakas efter godkännandet och möjliggör att samla in ytterligare uppgifter på ett strukturerat sätt. […].
– Noggrann övervakning av tillverkningen, inklusive av vaccinbatcher och dess distribution, med samma löpande kontroller som för alla godkända läkemedel. Övervakningen av tillverkningsprocesserna säkerställer att läkemedlet tillverkas och kontrolleras enligt höga farmaceutiska standarder vid storskalig kommersialisering.
Enligt EU:s villkorliga godkännande för försäljning är innehavaren av godkännandet för försäljning ansvarig. Innehavaren av godkännandet för försäljning ansvarar för produkten och dess säkra användning.
Detta låter fantastiskt i teorin. Det är vad Ursula lovade när hon personligen ringde och vred om armarna på politikerna i medlemsstaterna. Kanske var det inte ens nödvändigt att vrida armarna särskilt hårt eftersom politikerna var tillräckligt terroriserade av den covidpropagandan och väntade på att mirakelvaccinen skulle rädda dem. Problemet är att Ursula aldrig hade för avsikt att uppfylla dessa löften, och det är inte möjligt att producera mRNA-”vaccin” med den säkerhet, effektivitet och tillverkningskvalitet som krävs för läkemedel. Det som Ursula verkligen ville få ut av denna process var att binda samman alla EU:s medlemsstater i en pakt genom att lova en ”robust” CMA, så att ingen kunde ha en oberoende myndighet över de sprutor som distribuerades i deras länder. Artikel 5-systemet skulle ha inneburit att varje medlemsstat kunde godkänna produkten, och sedan ha befogenhet att återkalla godkännandet om några problem upptäcks. Artikel 5 ger också tillverkaren ansvarsfrihet, men gör det dock omöjligt att göra produkten obligatorisk och tvingande att ta. Med CMA:s väg skulle ingen av medlemsstaterna kunna utöva sitt oberoende beslutsfattande, och därför skulle Ursula kunna tvinga in dem alla i samma vansinniga och nästan komplett svartredigerade avtal med Pfizer, Moderna och AstraZeneca, som befriade bolagen från allt ansvar och dessutom förbjöd länderna att ändra sina egna lagar när det gäller ansvaret!
Köparna måste ”ersätta, försvara och hålla Pfizer skadeslös … från och mot alla stämningar, anspråk, åtgärder, krav, förluster, skador, skulder, förlikningar, straff, böter, kostnader och utgifter … som uppstår till följd av, är relaterade till, eller är en följd av vaccinet”
Noterbara större invändningar eller brist på invändningar från EMA:s granskare.
Avsnittet om kemisk tillverkning och kontroll (CMC) i ansökan om bioläkemedelslicens är den viktigaste pelaren i det regulatoriska godkännandet. Här beskrivs tillverkningsprocessen och efterlevnaden av god tillverkningspraxis (cGMP) och en omfattande uppsättning lagar och förordningar som är utformade för att garantera renhet, styrka, konsistens och säkerhet hos massproducerade läkemedel och biologiska läkemedel. Säkerhets- och effektdata från kliniska prövningar är värdelösa om tillverkaren inte kan försäkra tillsynsmyndigheterna och det medicinska samfundet om att: 1) att produkten i fråga enligt specifikationerna användes i kliniska prövningar, 2) att produkten är konsekvent tillverkad, ren, av hög kvalitet, reproducerbar, med väl karakteriserade och förutsägbara tillverkningsprocesser och kontrollsteg, 3) att samma produkt som prövades kommer att distribueras kommersiellt.
Det identifierades problem med CMC-avsnittet i Pfizers ansökan:

CMC-bedömarna var inte nöjda med den tidsram som gavs för bedömningen, eftersom den överskred alla normala tidsramar och även alla påskyndade tidsramar med stor marginal. Så lösningen var att utvärderarna bara behövde ”pressas”. På så sätt uppnåddes endast ett mål – att tvinga människor som eventuellt kunde ha uttryckt oro till gränsen till utmattning så att de helt enkelt gav upp och gick med på det. De högsta cheferna visste mycket väl att den rättsliga granskningen inte hade någon betydelse och ingen inverkan på det falska ”godkännandet”, utan att den skulle ske oavsett vad man gjorde. I Storbritannien har MHRA redan erkänt att de inte hade någon formell delegering av befogenhet att granska och godkänna dessa injektioner, och jag är villig att slå vad om att EMA inte heller hade någon sådan befogenhet.
CMC:s bedömare på lägre nivå visste inte detta och arbetade mycket hårt och troligen i god tro. I slutet av november hade de gjort över 140 formella invändningar mot Pfizers CMC-underlag, som fortfarande hade många gapande hål och saknade information. För referens så räcker normalt 10-15 invändningar från myndigheter för att en läkemedelsansökan stoppas från att gå vidare tills invändningarna är lösta. Tre större invändningar, dvs. formella röda flaggor, diskuteras särskilt i e-postmeddelandena nedan. Jag och andra har skrivit utförligt om MO#2 (bristande mRNA-integritet). Här finns ett e-postmeddelande från en av granskarna, Evdokia Korakianiti, och ett svar från Alexis Nolte som diskuterar problemet och dess inverkan (helt okänd och potentiellt mycket oroande) på produktens effektivitet och säkerhet:

Problemet med mRNA-nedbrytning diskuterades också av CMC-kvalitetsspecialisterna Ton van der Stappen, senior biofarmaceutisk expert vid Medicines Evaluation Board (baserat i Nederländerna) och kvalitetsspecialist för EMA:

och Brian Dooley, en annan kvalitetsspecialist på läkemedelsområdet vid EMA:

Bilderna nedan är från den kvalitetsgranskning som de har lämnat in till EMA. Den första bilden handlar om det nu väldokumenterade problemet med nedbrytning av mRNA i olika partier av Pfizers produkt. Här listas och färgkodas de analysresultat som Pfizer har tillhandahållit för de olika tillverkningsställena samt för läkemedelssubstansen och läkemedelsproduktkategorin. Läkemedelssubstans = produktens aktiva komponenter (enbart mRNA) och läkemedelsprodukt är den substans som är formulerad i lipiderna och andra ingredienser. % mRNA-integritet beskriver den procentuella andelen ”full längd” mRNA som upptäcktes i en sats. den andra delen av satsen bestod av okända trasiga bitar med okända egenskaper och okänd inverkan på säkerheten. Observera att tillsynsmyndigheterna inte utförde någon oberoende kontroll av något av detta, utan de listade helt enkelt de siffror som Pfizer/BioNTech tillhandahöll.
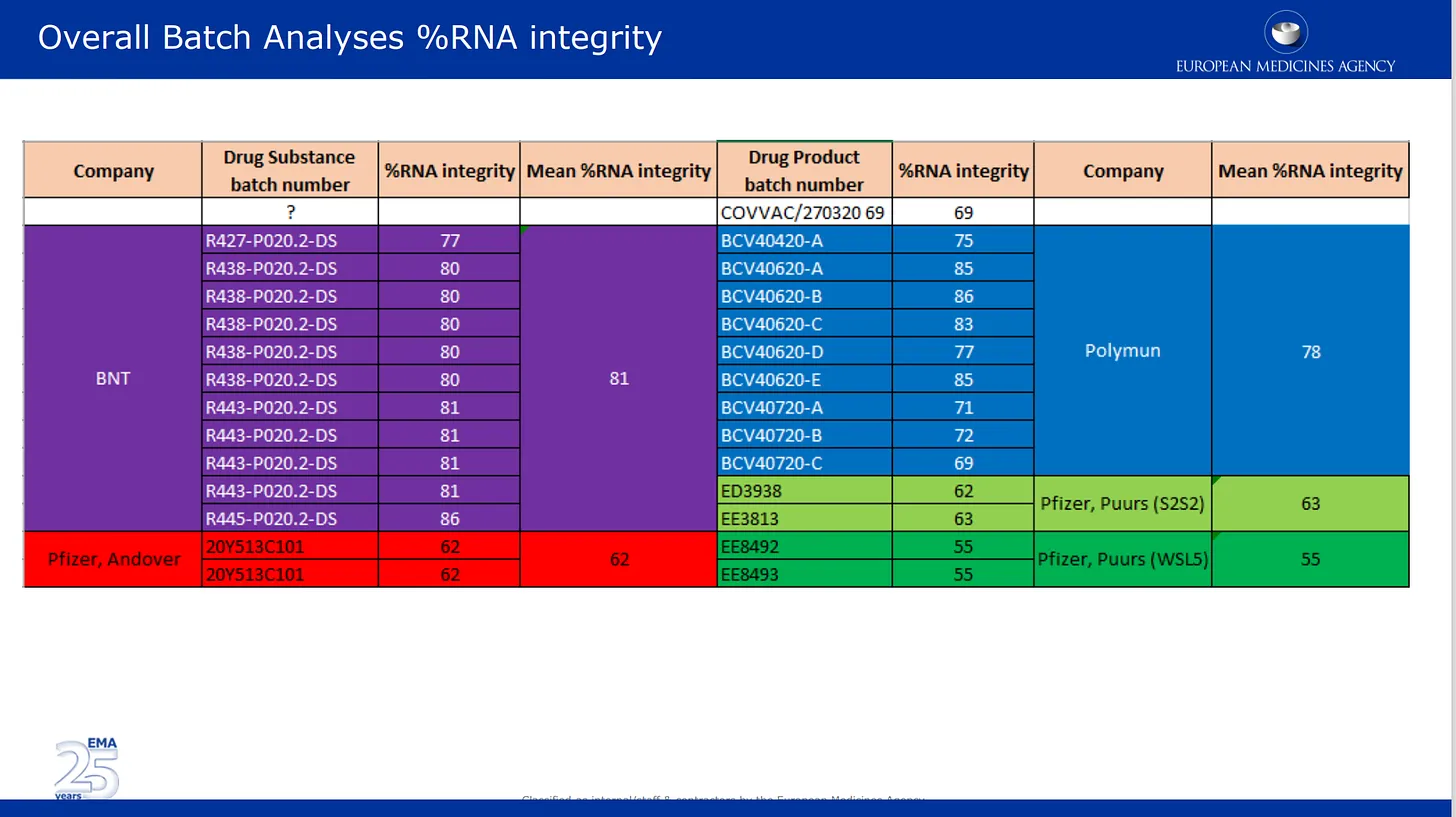
Det verkar som om dessa två vetenskapliga konsulter granskade och accepterade de falska bilderna av Western blot-resultaten som Pfizer lämnade in till EMA – här är de i deras egen PowerPoint-presentation från den 24 november 2020. Läs anteckningen under bilden – de accepterar dessa bilder som riktiga, trots att båda dessa granskare borde veta bättre. Varför protesterade de INTE mot detta? Så här står det i anteckningen:
Proteinstorleken efter in-vitro-uttryck av BNT162b2-läkemedelssubstansen bestämdes med hjälp av Western blot. Det bekräftades att den uttryckta proteinstorleken var jämförbar för de tre process 1-satserna och process 2-satsen. Figur 3.2.S.2.6-15 visar att den uttryckta proteinstorleken överensstämmer med den förväntade storleken på BNT162b2-läkemedelssubstansen och är jämförbar för alla testade tillverkningssatser. Dessutom är de relativa uttrycksnivåerna jämförbara för alla tillverkningssatser, vilket framgår av jämförbar bandintensitet vid varje belastningsnivå för alla batcher.

Kanske borde några hårdföra journalister kontakta dr van der Stappen och Dooley, och Korakianiti och andra personer som nämns här för att höra dem kommentera det här.
Det framgår tydligt av de svar som Evdokia Korakianiti tagit upp att EMA:s ledning la sig platt och förlitade sig på ”data som endast FDA har sett”, men är ”optimistisk” och att FDA hävdade att mRNA-brott var ett ”teoretiskt problem”. Är det sant? Finns det några uppgifter som stöder detta påstående eller inte? Här finns e-postmeddelanden som visar att de stora invändningarna formellt skrevs ner och sedan ignorerades av EMA eftersom produkten levererades kommersiellt bara några veckor senare. Villkoren i CMA uppfylldes aldrig.
Detta bekräftar vad vi redan vet – varken EMA (eller FDA, Health Canada, MHRA eller andra tillsynsmyndigheter) hade någon verklig auktoritet över dessa produkter eller någon inverkan på om de skulle användas på den intet ont anande allmänheten. Allt var teater från början till slut.
Här är de tre största invändningarna som fortfarande är olösta:

Och här är en del handuppräckning och ett godtagande av FDA:s påståenden utan att EMA:s tillsynsmyndigheter ifrågasätter eller gör någon formell utvärdering av data:

Vad jag kan säga avslutningsvis är att jag räknade till cirka 70 olika personer som nämns i det läckta dokumentet och i e-postmeddelandena och som tog del i denna tragiska charad – ”godkännandet” av den mest dödliga produkt som någonsin släppts lös på det största antalet människor, vilket resulterade i en aldrig tidigare skådad mängd döda och skadade i hela världen. Kanske, med några få undantag, blev de flesta av dem lurade 2020 och förstod inte att de deltog i ett krigsbrott och skrev under på ett dödligt bedrägeri. Jag tror att de flesta av dem vet nu och jag hoppas att de är tillräckligt förskräckta över vad de har möjliggjort, och jag hoppas att dessa människor träder fram som visselblåsare och börjar prata. Vi behöver svar.
Ursprungligen publicerad av Due Diligence and Art
Suggest a correction




