Cartas do Submundo
Crédito da imagem: Shutter Stock
Revisitados os e-mails da EMA que vazaram
No final de 2020, uma colecção de documentos – cerca de 900 páginas da secção de fabrico e controlo químico (CMC) da “vacina” da Pfizer – da submissão regulamentar à Agência Europeia de Medicamentos (EMA) foi divulgada e enviada a vários jornalistas. Os documentos incluíam também trocas de correio electrónico entre alguns dos revisores e quadros superiores da EMA. A fuga de informação foi objecto de cobertura pelo British Medical Journal, que conseguiu confirmar a autenticidade dos documentos. Recebi estes documentos de um colega cerca de um ano depois, no final de 2021, e li e utilizei muitos deles nas minhas análises. No meu trabalho profissional, vi muitos documentos da Pfizer relacionados com I&D, pelo que também posso confirmar que estes documentos que vazaram eram altamente consistentes com a documentação típica da Pfizer. A EMA não negou a autenticidade e apenas afirmou que os cabeçalhos de alguns dos e-mails foram alterados.
Tenho agora um número muito maior de seguidores e uma compreensão muito mais clara da organização e da estrutura pseudo-legal do cartel criminoso que está a conduzir a atrocidade global coloquialmente conhecida como a “resposta à pandemia de covid”. Estou a revisitar os e-mails que vazaram, pois acredito que eles fornecem algumas provas altamente significativas.
Os ficheiros de correio electrónico da EMA que li contêm 14 capturas de ecrã de mensagens de correio electrónico de meados a finais de Novembro de 2020. As trocas de mensagens são do pessoal da EMA e de executivos seniores. Na minha opinião, estas mensagens de correio electrónico demonstram o seguinte:
- Os revisores da EMA estavam sob uma enorme pressão política para inventar novas formas de aprovar os produtos perigosos não aprováveis. A pressão estava a emanar do topo dos governos dos EUA, do Reino Unido e da UE.
- A Comissária da UE, Ursula von der Leyen, fez promessas aos Estados-Membros que nunca tencionou cumprir, a fim de os vincular a todos num único pacto para os contratos de vacinas e, assim, evitar quaisquer decisões independentes nos seus próprios países.
- Houve problemas graves e insolúveis – dado o calendário propositadamente nada realista – com a qualidade do produto que o pessoal da EMA foi pressionado a aprovar. Alguns não se sentiram à vontade para o fazer e manifestaram as suas preocupações. Outros “ignoraram” dados claramente inventados.
Em última análise, a revisão regulamentar em si e as preocupações levantadas não tiveram importância – o produto ia ser comercializado de qualquer forma. Agora sabemos exactamente porquê – as autoridades reguladoras não tinham poder de regulamentação sobre o produto. Os reguladores farmacêuticos não supervisionam os materiais militares conhecidos como “contramedidas” e “demonstrações de fabrico” (uma linguagem tímida que encobre os agentes de guerra biológica fabricados pelo Governo dos EUA capturado e pelos seus parceiros globais). Os e-mails mostram que a maioria dos funcionários da EMA foram actores involuntários nesta peça.
A confirmação deste facto para o Reino Unido foi divulgada recentemente.
Com base na resposta à MHRA FOIA:
“Todas as vacinas Covid e as decisões de autorização terapêutica foram tomadas pelo Ministro do Licenciamento e não foram delegadas.”
Tradução – normalmente, a autoridade para analisar e aprovar novos produtos farmacêuticos é formalmente delegada à MHRA pelo Secretário de Estado da Saúde (Reino Unido). No caso dos produtos da covid, a delegação de autoridade não existe. Ao que parece, todos eles foram implementados por Matt Hancock sozinho (embora ele esteja a apontar o dedo a alguém superior). A mesma coisa aconteceu nos EUA – Alex Azar, sob a administração Trump, implantou esses biomateriais não conformes nos americanos, e Xavier Becerra, sob Biden, continua a fazê-lo hoje.
A pressão política.
A seguinte troca de e-mails ocorreu em 16 de novembro de 2020 entre executivos seniores da EMA:
Noel Wathion – Diretor Executivo Adjunto (aposentado em junho de 2021):

Agnes Saint-Raymond – Chefe da Divisão de Assuntos Internacionais:

Marco Cavaleri, Presidente do Grupo de Trabalho sobre a Pandemia de Covid-19 da EMA:

Os e-mails devem ser lidos de baixo para cima.

Algumas coisas interessantes: os três reguladores – FDA dos EUA, MHRA do Reino Unido e EMA da UE – estão todos ocupados a coordenar o calendário de aprovação antes de qualquer revisão formal dos dados, antes de os comités consultivos terem visto os resultados dos ensaios clínicos, os terem discutido, votado sobre eles, etc. Estão a discutir o calendário, uma vez que os dados NÃO IMPORTAM para saber se estes produtos vão ser comercializados ou não. Além disso, estão a interagir como se não fossem três agências separadas de nações soberanas distintas, responsáveis perante conjuntos separados de contribuintes e de supervisão congressional/parlamentar, mas simplesmente departamentos burocráticos já fundidos num governo global. Por fim, a FDA vai “apressar-se a entrar nos EUA”, sendo “empurrada por Azar” (Alex Azar – Secretário do HHS na altura) e “Trump está a puxar os cordelinhos”.
Muitas pessoas perguntam-me como é possível que milhares de pessoas tenham participado na fraude orquestrada como “resposta à pandemia de covid” – certamente não é possível ter tantas pessoas em conluio! Não era necessário ter tantos a saber. Neste caso, Noel Wathion, um executivo de topo da EMA, ou não está ciente de que a análise dos dados é irrelevante para a colocação das injecções no mercado, ou deturpou habilmente este facto (na verdade, acredito que ele não estava ciente). Por conseguinte, o pessoal da EMA abaixo dele não precisaria de estar ciente e seria simplesmente apressado a fazer qualquer tarefa limitada que lhe fosse atribuída. A compartimentação é a chave para encobrir qualquer fraude importante no seio de grandes organizações e estruturas complexas. Terá sido por isso que se demitiu/reformou pouco depois do lançamento dos kill shots? Ele também está sob pressão da CE (Comissão Europeia) para aprovar. E a Pfizer quer agora uma Autorização de Introdução no Mercado (AIM) completa em vez da Condicional (ACM)! Nota – a CMA foi emitida, mas as condições nunca foram cumpridas pela Pfizer/BioNTech, porque quem é que se importa, era um jogo desde o início.
“(Co)-Rapps” = co-relatores. A EMA é uma entidade europeia, composta pelas “autoridades competentes” dos Estados-Membros, anteriormente separadas, que costumavam regulamentar e aprovar os produtos farmacêuticos em cada país separadamente. Na estrutura europeia, a equipa de análise técnica e de co-análise é seleccionada para um produto específico. No caso das “vacinas” contra a COVID-19, a equipa sueca, chefiada por Philip Josephson, foi a relatora (revisor principal) e a equipa francesa, chefiada por Jean-Michel Race, foi a co-relatora. “CHMP” =Comité dos Medicamentos para Uso Humano (na EMA).

O e-mail é dirigido a Olga Solomon na CE (Comissão Europeia), e a chefe de Noel, Emer Cooke – Directora Executiva da EMA e antigo quadro superior da OMS – é copiado. Aqui está Emer Cooke:

O Pacto Inteligente de Ursula.
Lembram-se dela?

Ursula von der Leyen – Comissária da UE, cujos feitos incluem a negociação de incríveis contratos predatórios de fornecimento da Pfizer em nome de todos os Estados-Membros da UE através de mensagens de texto com o Director Executivo da Pfizer, Albert Bourla. Nestes contratos, os países da UE tiveram de colocar bens do Estado como garantia, renunciar a todas as leis de controlo de qualidade, de importação e de protecção dos consumidores e abdicar da soberania nacional – ou seja, não foram autorizados a alterar a legislação relativa à responsabilidade pelas vacinas pelos seus próprios parlamentos? Os contratos predatórios que foram completamente redigidos para proteger os chamados “interesses comerciais da Pfizer”. A seguinte troca de correio electrónico está relacionada com os valentes esforços de Ursula:

Há uma série de acrónimos utilizados, os mais relevantes são “CE” = Comissão Europeia, “EM” = Estados-Membros, “PE” = Parlamento Europeu. A frase-chave é que Ursula está “disposta a contactar pessoalmente os ministros da saúde competentes para evitar a utilização do nº 2 do artigo 5º”. De que se trata? O n.º 2 do artigo 5.º refere-se ao “n.º 2 do artigo 5.º da Directiva 2001/83” – autorização de utilização de emergência num Estado-Membro europeu, concedida por cada um dos Estados-Membros separadamente nos seus próprios países. A CMA é uma autorização condicional de introdução no mercado emitida pela EMA para todos os membros da UE em simultâneo. Anunciada pela UE como um processo muito mais sólido do que uma AIM (sublinhado meu):
…a CMA segue um quadro controlado e sólido que oferece salvaguardas que as autorizações de utilização de emergência podem não oferecer. Na realidade, uma autorização de utilização de emergência não é uma autorização da vacina, mas uma autorização da utilização temporária da vacina não autorizada. A CMA assegura que todos os controlos de farmacovigilância e de fabrico, incluindo controlos de lotes para vacinas e outras obrigações pós-aprovação, se aplicam de forma juridicamente vinculativa […]. Nomeadamente:
-Garante um controlo rigoroso, através do sistema de farmacovigilância da UE, da segurança do medicamento em toda a UE. […]
-Garante a monitorização da segurança pós-autorização e permite a recolha de dados adicionais de forma estruturada. […].
-O fabrico rigoroso, incluindo a libertação de lotes para vacinas e distribuição, está sujeito aos mesmos controlos contínuos que para todos os medicamentos autorizados. A monitorização dos processos de fabrico garante que o medicamento é fabricado e controlado de acordo com elevados padrões farmacêuticos no contexto da comercialização em grande escala.
Ao abrigo de uma Autorização de Introdução no Mercado Condicional (ACM) da UE, a responsabilidade recai sobre o titular da autorização de introdução no mercado. O titular da autorização de introdução no mercado será responsável pelo produto e pela sua utilização segura.
Em teoria, isto parece óptimo. Foi o que Ursula prometeu quando telefonou pessoalmente e torceu os braços aos políticos dos Estados-Membros. Talvez o braço-de-ferro nem fosse necessário, uma vez que estavam suficientemente aterrorizados com a propaganda da covid e à espera que as “vacinas” milagrosas os salvassem. O problema é que Ursula nunca teve a intenção de cumprir estas promessas e, de qualquer modo, não é possível produzir as “vacinas” de mRNA com a segurança, a eficácia e a qualidade de fabrico exigidas aos produtos farmacêuticos. O que Ursula realmente pretendia com este processo era unir todos os Estados-Membros europeus num pacto, prometendo uma CMA “robusta”, para que não pudessem ter uma autoridade independente sobre as vacinas distribuídas nos seus países. A via do artigo 5.º teria significado que cada Estado-Membro poderia autorizar o produto, tendo depois o poder de revogar a autorização se fossem detectados problemas. O artigo 5.º também prevê uma isenção de responsabilidade para o fabricante, mas impossibilita a autorização do produto. Com a via da CMA, nenhum dos Estados-Membros poderia exercer o poder de decisão independente, pelo que a Comissária poderia forçá-los a todos a assinar os mesmos contratos insanos e quase totalmente redigidos da Pfizer, da Moderna e da AstraZeneca, que, de qualquer modo, renunciavam a toda a responsabilidade e proibiam ainda os países de alterar as suas próprias leis em matéria de responsabilidade!
Os compradores devem “indemnizar, defender e isentar a Pfizer … de e contra todos e quaisquer processos, reclamações, acções, exigências, perdas, danos, responsabilidades, acordos, penalidades, multas, custos e despesas … decorrentes de, relacionados com, ou resultantes da Vacina”
Principais objecções notáveis e falta delas por parte dos revisores da EMA.
A secção de fabrico e controlos químicos (CMC) do pedido de licença biológica é o principal pilar da aprovação regulamentar. Descreve o processo de fabrico e a conformidade com as Boas Práticas de Fabrico (BPF), bem como um vasto conjunto de leis e regulamentos destinados a garantir a pureza, a potência, a consistência e a segurança dos medicamentos e produtos biológicos produzidos em massa. Os dados de segurança e eficácia dos ensaios clínicos são inúteis se o fabricante não puder garantir às entidades reguladoras e à comunidade médica que 1) o produto em questão, de acordo com as especificações, foi utilizado em ensaios clínicos, 2) o produto é fabricado de forma consistente, puro, de alta qualidade, reprodutível, com um processo de fabrico e etapas de controlo bem caracterizados e previsíveis, 3) o mesmo produto que foi testado será distribuído comercialmente.
Foram identificados problemas na secção CMC da apresentação da Pfizer:

Os avaliadores do CMC não ficaram satisfeitos com o calendário apresentado para a avaliação, uma vez que este violava todos os prazos normais e todos os prazos acelerados, também, por uma grande margem. Por isso, a solução foi que os avaliadores só precisavam de ser “empurrados”. Isto só atingiu um objectivo – forçar as pessoas que poderiam potencialmente ter levantado preocupações até à exaustão, de modo a que simplesmente desistissem e alinhassem. Afinal de contas, os responsáveis máximos sabiam bem que a revisão regulamentar não tinha qualquer significado nem implicação na falsa “aprovação”, que iria acontecer de qualquer forma. No Reino Unido, a MHRA já admitiu que não tinha delegação formal de autoridade para rever e aprovar estas injecções, e estou disposto a apostar que a EMA também não tinha essa autoridade.
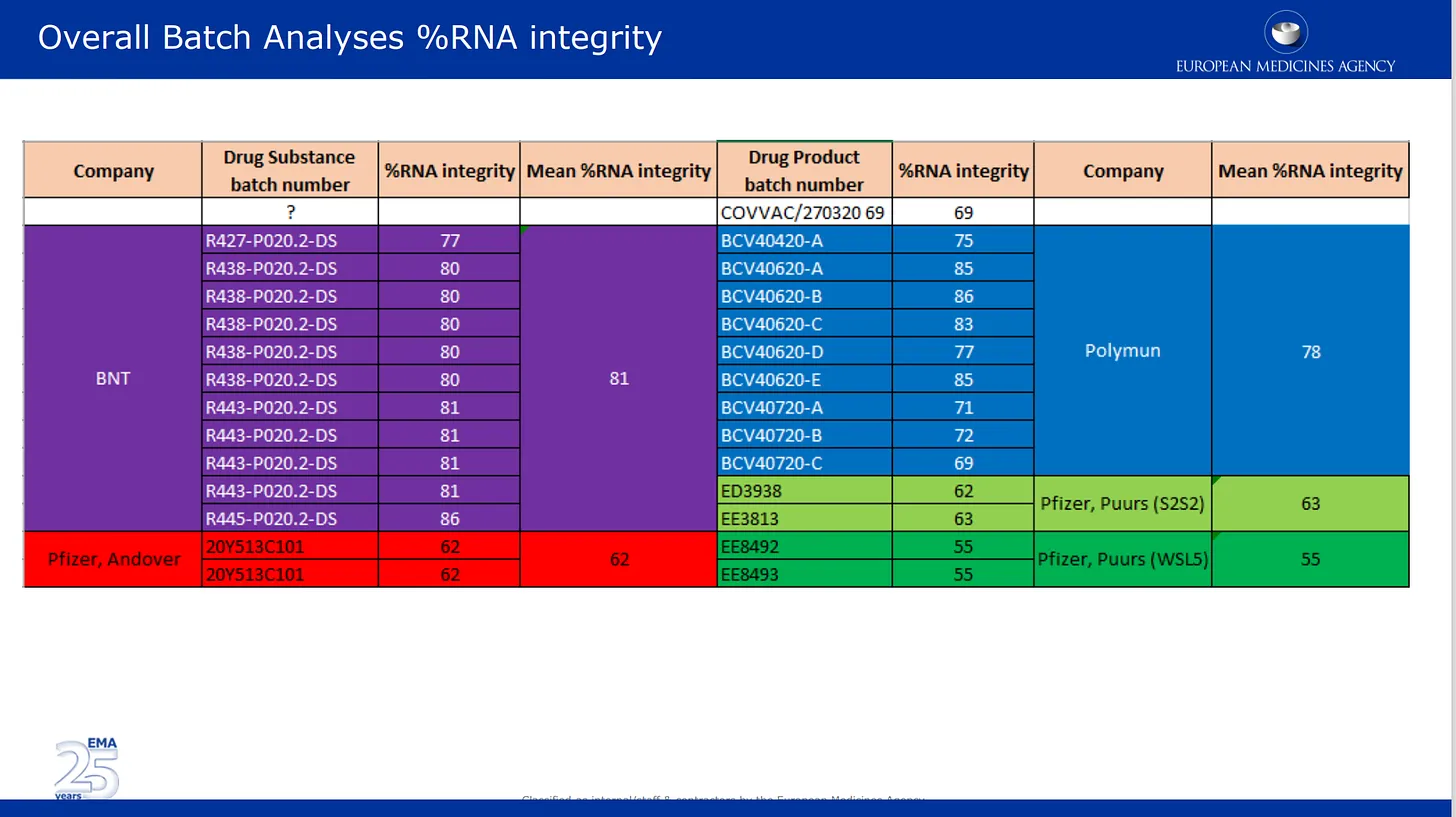
Os avaliadores de baixo nível do CMC não sabiam disso e estavam a trabalhar arduamente e, muito provavelmente, de boa fé. No final de Novembro, levantaram mais de 140 objecções formais à apresentação do CMC da Pfizer, que ainda tinha muitas lacunas e informações em falta. Para referência, 10-15 objecções regulamentares normalmente impedem que um pedido farmacêutico avance até que as objecções sejam resolvidas. Três Objecções Principais, ou seja, bandeiras vermelhas formais, são discutidas especificamente nos e-mails abaixo. Eu e outros escrevemos extensivamente sobre a MO#2 (falta de integridade do mRNA). Aqui está uma mensagem de correio electrónico de um dos revisores, Evdokia Korakianiti, e uma resposta de Alexis Nolte que discute o problema e o impacto (completamente desconhecido e potencialmente muito preocupante) na eficácia e segurança do produto:

O problema da degradação do ARNm foi também discutido pelos especialistas em qualidade CMC Ton van der Stappen, perito biofarmacêutico sénior do Conselho de Avaliação de Medicamentos (com sede nos Países Baixos) e especialista em qualidade da EMA:

e Brian Dooley, outro especialista em qualidade farmacêutica da EMA:

As imagens abaixo são da revisão de qualidade apresentada por eles à EMA. A primeira imagem fala sobre a questão, agora bem documentada, da degradação do mRNA em diferentes lotes do produto da Pfizer. Aqui, os resultados da análise de lotes fornecidos pela Pfizer são listados e codificados por cores pelos diferentes locais de fabrico, bem como por substância medicamentosa e categoria de medicamento. Substância medicamentosa = componentes activos do produto (mRNA isolado) e produto medicamentoso é a substância formulada nos lípidos e outros ingredientes. A % de integridade do mRNA descreve a % de mRNA de “comprimento total” detectada num lote. A outra parte do lote era composta por fragmentos desconhecidos com propriedades ou impacto desconhecidos na segurança. Note-se que os reguladores não efectuaram qualquer verificação independente de nenhum destes elementos, limitando-se a enumerar os números fornecidos pela Pfizer/BioNTech.
Parece que estes dois consultores científicos reviram e aceitaram as imagens falsas dos resultados do Western blot apresentados pela Pfizer à EMA – aqui estão eles na sua própria apresentação PowerPoint de revisão de 24 de Novembro de 2020. Leia a nota sob o slide – eles estão aceitando essas imagens como reais, embora ambos os revisores devam saber melhor. Porque é que eles NÃO se opuseram a isto? Isto é o que diz a nota:
O tamanho da proteína após a expressão in-vitro da substância medicamentosa BNT162b2 foi determinado utilizando Western blot. O tamanho da proteína expressa foi confirmado como sendo comparável para três lotes do Processo 1 e para o lote do Processo 2. A Figura 3.2.S.2.6-15 mostra que o tamanho da proteína expressa é consistente com o tamanho esperado da substância medicamentosa BNT162b2 e comparável em todos os lotes testados. Além disso, os níveis de expressão relativa são comparáveis para todos os lotes, como evidenciado pela intensidade comparável da banda em cada nível de carga em todos os lotes.

Talvez alguns jornalistas de grande impacto devessem contactar os Drs. van der Stappen e Dooley, bem como a Sra. Korakianiti e outros indivíduos aqui mencionados, para obterem comentários.
É evidente, pelas respostas dadas por Evdokia Korakianiti, que a direcção da EMA prescindiu de armas e se baseou em “dados que só a FDA viu”, mas é “optimista” e que a FDA afirmou que a quebra do mRNA era uma “preocupação teórica”. A sério? Há dados que sustentem esta afirmação ou não? Aqui estão os e-mails que indicam que as objecções principais foram formalmente redigidas e subsequentemente ignoradas pela EMA, uma vez que o produto foi comercializado apenas algumas semanas mais tarde. As condições da CMA nunca foram cumpridas.
Isto confirma o que já sabíamos – nem a EMA (nem a FDA, a Health Canada, a MHRA ou outras entidades reguladoras) tinha qualquer autoridade real sobre estes produtos ou impacto sobre a sua utilização junto de um público desprevenido. Foi tudo teatro do princípio ao fim.
Aqui estão as três principais objecções que continuam por resolver até à data:

E aqui estão alguns acenos de mão e a aceitação das afirmações da FDA sem qualquer questionamento ou avaliação formal dos dados por parte dos reguladores da EMA:

O que posso dizer para terminar – contei cerca de 70 pessoas diferentes mencionadas no documento que vazou e nos e-mails, que facilitaram esta trágica charada – a “aprovação” do produto mais letal alguma vez lançado sobre o maior número de pessoas, resultando num número de mortos e feridos sem precedentes em todo o mundo. Talvez, com algumas excepções, a maioria deles tenha sido enganada em 2020 e não tenha compreendido que estava a participar num crime de guerra e a assinar uma fraude mortal. Acredito que a maioria deles já sabe, espero que estejam suficientemente horrorizados com o que permitiram, e espero que essas pessoas se apresentem como denunciantes e comecem a falar. Precisamos de respostas.
Originalmente publicado por Due Diligence and Art
Suggest a correction