Letters from the Underworld
Image credit: Shutter Stock
Leaked Emails from EMA Revisited
In late 2020 a collection of documents – approximately 900 pages from Pfizer’s “vaccine” Chemistry Manufacturing and Controls (CMC) section of the regulatory submission to the European Medicines Agency (EMA) were leaked and sent to a number of journalists. The documents also included email exchanges from some of the reviewers and senior executives at the EMA. The leak was covered in the British Medical Journal who were able to confirm that the documents were authentic. I received these documents from a colleague about a year later, at the end of 2021, and I read and used a lot of them in my analyses. I have seen many R&D related documents from Pfizer in my professional work, so I can also confirm that these leaked ones were highly consistent with typical Pfizer documentation. The EMA did not deny the authenticity, and only stated that the headers of some of the emails were changed.
I have a much larger number of followers now and a much clearer understanding of the organization and pseudo-legal structure of the criminal cartel driving the global atrocity colloquially known as the “covid pandemic response”. I am revisiting the leaked emails as I believe that they provide some highly significant evidence.
The EMA email files that I have read contain 14 screenshots of emails from mid to late November 2020. The exchanges are from the EMA staff and senior executives. In my opinion these emails demonstrate that:
- The EMA reviewers were under a massive political pressure to invent new ways of approving the unapprovable dangerous products. The pressure was emanating from the very top of the US, UK and EU governments.
- EU Commissioner, Ursula von der Leyen made promises to the Member States she was never intending to fulfill in order to tie them all in a single pact for vaccine contracts and thus pre-empt any independent decisions in their own countries.
- There were severe and unresolvable – given the purposefully unrealistic timeline – issues with quality of the product the EMA staff were pressured to OK. Some were uncomfortable with doing so and voicing their concerns. Others “overlooked” clearly made-up data.
Ultimately, the regulatory review itself and the concerns raised did not matter – the product was going to be marketed regardless. We now know exactly why – the regulatory authorities did not have the regulatory power over it. The pharmaceutical regulators do not oversee military materials known as “countermeasures” and “manufacturing demonstrations” (coy language covering up the biowarfare agents made by the captured US Government and its global partners). The emails show that the majority of the EMA staff were unwitting actors in this play.
Confirmation of this for the UK came out recently.
Based on the response to MHRA FOIA:
“All the Covid vaccines and therapeutics authorisation decisions were taken by the Licensing Minister and were not delegated.”
Translation – normally the authority to review and approve new pharmaceuticals is formally delegated to the MHRA from the Secretary of State for Health (UK). In the case of covid products, the delegation of authority does not exist. It appears that all of them were deployed single-handedly by Matt Hancock (although he is pointing the finger at someone higher up). The same thing happened in the US – Alex Azar under the Trump Administration deployed these non-compliant biomaterials on Americans, and Xavier Becerra under Biden continues to do so today.
The Political Pressure.
The following email exchange occurred on November 16, 2020 between senior executives of the EMA:
Noel Wathion – Deputy Executive Director (retired in June 2021):

Agnes Saint-Raymond – Head of the International Affairs Division:

Marco Cavaleri, Chair of the Covid-19 Pandemic Task Force at EMA:

The emails should be read from bottom to top.

A couple of interesting things: the three regulators – US FDA, UK MHRA and EU EMA – are all busy coordinating the timing of approval before any formal review of the data has happened, before advisory committees have seen the clinical trial results, discussed them, voted on them, etc. They are discussing the timing as the data DO NOT MATTER as to whether these products are going on the market or not. In addition, they are interacting as if they are not three separate agencies of separate sovereign nations responsible to separate sets of taxpayers and congressional/parliamentary oversight, but simply bureaucratic departments already merged into one global government. Finally, the FDA is going to “rush into EUA”, being “pushed by Azar” (Alex Azar – HHS Secretary at the time) and “Trump is pulling strings”.
Many people ask me how is it possible that thousands of people participated in the scam orchestrated as “covid pandemic response” – surely it’s not possible to have so many people in cahoots! It wasn’t necessary to have so many in the know. Here, Noel Wathion, a top EMA executive is either not aware that the data review is irrelevant to whether the injections would ultimately go on the market, or skillfully misrepresenting this (I in fact believe he was not aware). Therefore, the EMA staff below him would not need to be aware, and simply be rushed to do whatever narrow task they were assigned. Compartmentalization is key to covering up any major scam within large organizations and complex structures. Is this why he resigned/retired shortly after the rollout of the kill shots? He is also under EC (European Commission) pressure to approve. And Pfizer now wants a full Marketing Authorizations (MA) instead of the Conditional one (CMA)! Note – the CMA was issued, but the conditions were never fulfilled by Pfizer/BioNTech, because who cares, it was a game from the start.
“(Co)-Rapps” = co-rapporteurs. The EMA is a European entity, composed of Member States’ formerly separate “competent authorities” who used to regulate and approve pharmaceuticals in each country separately. In the European structure, the technical review and co-review team is selected for a specific product. In the case of the covid “vaccines”, the Swedish team headed by Philip Josephson was the Rapporteur (lead reviewer) and the French team headed by Jean-Michel Race – the co-rapporteur. “CHMP” =Committee for Medicinal Products for Human Use (at EMA).

The email is addressed to Olga Solomon at EC (European Commission), and Noel’s boss, Emer Cooke -Executive Director of EMA and former senior executive at WHO is copied. Here is Emer Cooke:

Ursula’s Clever Pact.
Remember her?

Ursula von der Leyen – EU Commissioner, whose achievements include negotiating incredible predatory Pfizer supply contracts on behalf of all EU Member States by text messages with Pfizer CEO Albert Bourla. In these contracts the EU countries had to put up state assets as collateral, waive all quality control, importation and consumer protection laws and give up national sovereignty – i.e., not allowed to change legislation with respect to vaccine liability by their own parliaments? The predatory contracts that were completely redacted to protect so-called “Pfizer commercial interests”. The following email exchange relates to Ursula’s valiant efforts:

There are a bunch of acronyms used, the most relevant are “EC” = European Commission, “MS” = Member States, “EP”=European Parliament. The key sentence is that Ursula is “prepared to call relevant health ministers personally to avoid the use of Art 5(2)”. What is this about? Art 5(2) refers to “Article 5(2) of Directive 2001/83” – emergency use authorization in a European Member State, given by each of the Member States separately in their own countries. CMA is a Conditional Market Authorization which is issued by the EMA for all EU members simultaneously. Advertised by the EU as a far more robust process than an EUA (emphasis added by me):
…CMA follows a controlled and robust framework providing safeguards that emergency use authorisations might not. In reality, an emergency use authorisation is not an authorisation of the vaccine but an authorisation of the temporary use of the unauthorised vaccine. The CMA ensures that all pharmacovigilance, manufacturing controls including batch controls for vaccines and other post-approval obligations apply in a legally binding manner […]. Notably:
•It ensures a rigorous monitoring, through the EU pharmacovigilance system, of the safety of the medicine across the EU. […]
•It ensures post-authorisation safety monitoring and allows the collection of additional data in a structured manner. […].
•Rigorous manufacturing including batch release for vaccines and distribution, are subject to the same ongoing controls as for all authorised medicines. The monitoring of the manufacturing processes ensures that the medicine is manufactured and controlled according to high pharmaceutical standards in the context of large scale commercialisation.
Under an EU Conditional Marketing Authorisation (CMA), liability is with the holder of the marketing authorisation. The marketing authorisation holder will be responsible for the product and its safe use.
This sounds terrific in theory. This is what Ursula promised when she personally called and twisted arms of politicians in the Member States. Maybe the arm-twisting wasn’t even necessary as they were sufficiently terrorized by the covid propaganda and awaiting the miracle “vaccines” to save them. The problem is that Ursula never meant to fulfill these promises, and at any rate, it is not possible to produce the mRNA “vaccines” to the safety, efficacy and manufacturing quality required of pharmaceuticals. What Ursula really needed from this process was to tie all the European Member States together in a pact by promising a “robust” CMA, so that they could not have an independent authority over the shots distributed in their countries. Article 5 path would have meant each MS could authorize the product, and would then have the power to revoke the authorization if any problems detected. Article 5 also provides a liability waiver to the manufacturer, however makes it impossible to mandate the product. With the CMA route none of the MSs could exercise the independent decision making, and so she would be able to then force them all into the same, insane and almost completely redacted Pfizer, Moderna and AstraZeneca contracts, which waived all liability anyway, and further prohibited the countries to change their own laws in regard to the liability!
Purchasers must “indemnify, defend and hold harmless Pfizer … from and against any and all suits, claims, actions, demands, losses, damages, liabilities, settlements, penalties, fines, costs and expenses … arising out of, relating to, or resulting from the Vaccine.”
Notable Major Objections and Lack Thereof by the EMA Reviewers.
Chemistry Manufacturing and Controls (CMC) section of the Biologics License Application is the main pillar of the regulatory approval. It describes the manufacturing process and compliance with the Good Manufacturing Practices (cGMP), and extensive set of laws and regulations designed to assure purity, potency, consistency and safety of the mass produced drugs and biologics. The safety and efficacy data from clinical trials are useless if the manufacturer cannot assure the regulators and medical community that: 1) the product in question per specification, was used in clinical trials, 2) product is consistently made, pure, high quality, reproducible, with well characterized and predictable manufacturing process and control steps, 3) the same product as was trialed will be commercially distributed.
There were problems identified with CMC section of Pfizer’s submission:

The CMC assessors were not happy with the timeline given for assessment, since it violated all the normal timelines and all the accelerated ones, too, by a large margin. So the solution was that the assessors just needed to be “pushed”. This accomplished only one goal – to force people who could have potentially raised concerns to the brink of exhaustion so that they simply give up and go along. After all, those at the top knew well that the regulatory review had no meaning and no implication on the fake “approval”, it was going to happen no matter what. In the UK, MHRA already admitted that they had no formal delegation of authority to review and approve these injections, and I am willing to bet that EMA did not have such authority either.
The CMC low level assessors did not know this, and were working very hard and most likely in good faith. As of late November, they raised 140+ formal objections to Pfizer’s CMC submission, which still had many gaping holes and missing information. For reference 10-15 regulatory objections normally stop a pharmaceutical application from going forward until objections are resolved. Three Major Objections, i.e. formal red flags are discussed specifically in the emails below. I and others wrote extensively about MO#2 (lack of mRNA integrity). Here is an email from one of the reviewers, Evdokia Korakianiti and a response from Alexis Nolte discussing the problem and the impact (completely unknown and potentially very troubling) on the efficacy and safety of the product:

The mRNA degradation problem was also discussed by the CMC Quality specialists Ton van der Stappen, Senior Biopharmaceutical Expert at Medicines Evaluation Board (based in the Netherlands), and Quality Specialist for the EMA:

and Brian Dooley, another Pharmaceutical Quality Specialist at EMA:

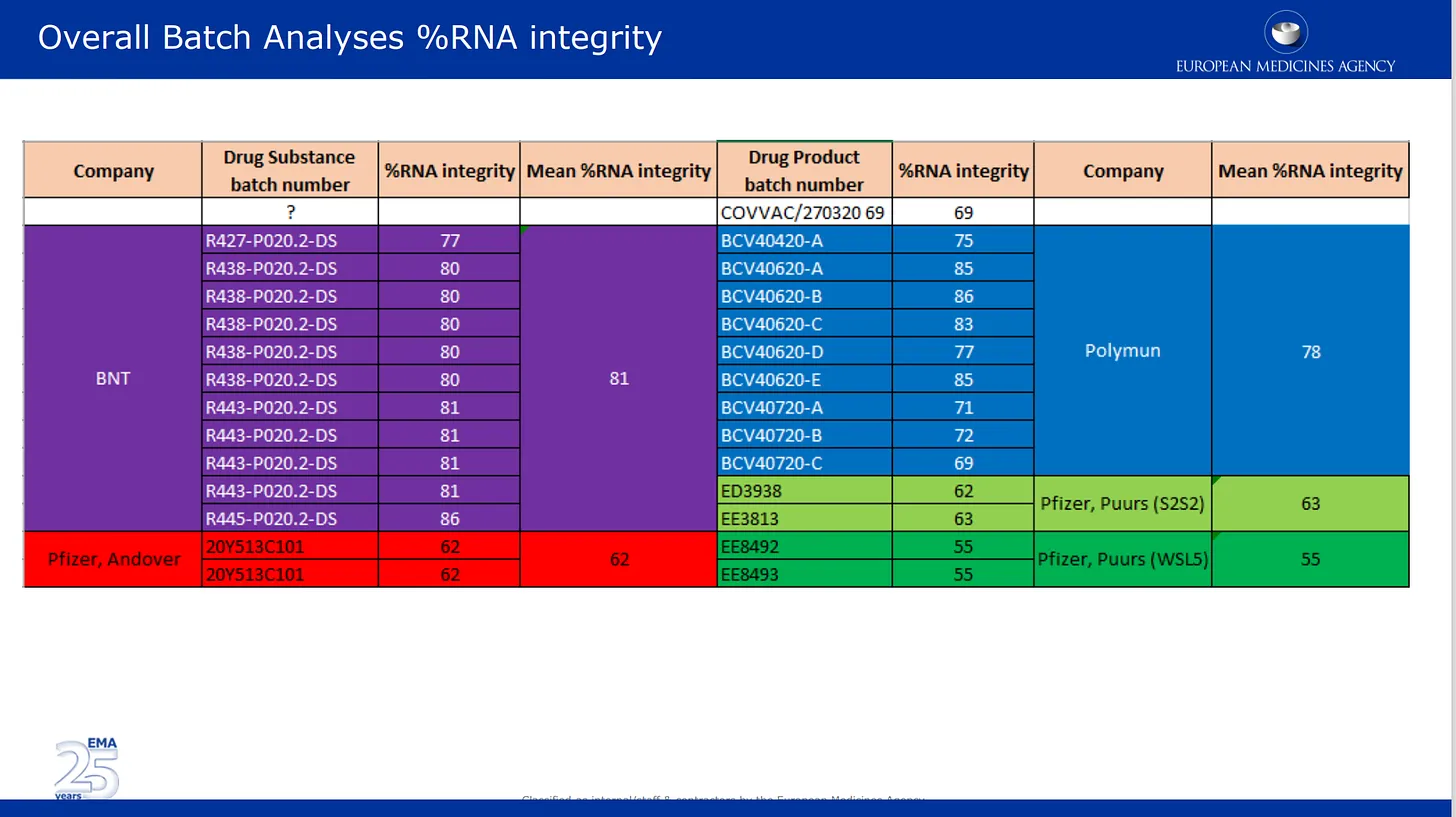
The images below are from the quality review submitted by them to the EMA. The first image talks about now well documented issue of the mRNA degradation in different batches of Pfizer’s product. Here the batch analysis results supplied by Pfizer are listed and color coded by the different manufacturing locations, as well as by drug substance and drug product category. Drug substance = active components of the product (mRNA alone) and drug product is the substance formulated in the lipids and other ingredients. The %mRNA integrity describes the % of “full length” mRNA detected in a batch. the other portion of the batch was composed of unknown broken pieces with unknown properties or impact on safety. Note that no independent check of any of this was performed by the regulators, they simply listed the numbers provided by Pfizer/BioNTech.
It appears that these two scientific consultants reviewed and accepted the fake images of the Western blot results submitted by Pfizer to the EMA – here they are in their own review PowerPoint presentation from November 24, 2020. Read the note under the slide – they are accepting these images as real, even though both of these reviewers should know better. Why did they NOT object to this? This is what the note says:
The protein size after in-vitro expression of BNT162b2 drug substance was determined using Western blot. Expressed protein size was confirmed to be comparable for three Process 1 batches and the Process 2 batch. Figure 3.2.S.2.6-15 shows that the expressed protein size is consistent with the expected size of BNT162b2 drug substance and comparable across all tested batches. In addition, relative expression levels are comparable for all batches, as evidenced by comparable band intensity at each load level across all batches.

Perhaps some hard-hitting journalists should reach out to Drs. van der Stappen and Dooley, and Ms. Korakianiti and other individuals mentioned here for comment.
It is evident from the responses raised by Evdokia Korakianiti the EMA management waived arms and relied on “data that only FDA has seen”, but is “optimistic” and that FDA claimed mRNA breakage was a “theoretical concern”. Really? Is there any data supporting this statement or nah? Here are the emails indicating that the Major Objections were formally written up and subsequently disregarded by the EMA since the product was shipped commercially just a couple of weeks later. The conditions of the CMA were never fulfilled.
This confirms what we already know – neither the EMA (nor the FDA, Health Canada, MHRA or other regulators) had any real authority over these products or impact on whether they were going to be deployed on unsuspecting public. It was all theater from start to finish.
Here are the three Major Objections that remain unresolved to date:

And here is some hand waving and taking the FDA’s assertions without any questioning or formal evaluation of data by the EMA regulators:

What I can say in closing – I counted approximately 70 different individuals mentioned throughout the leaked document and emails, who facilitated this tragic charade – the “approval” of of the most lethal product ever unleashed on the largest number of people, resulting in unprecedented death and injury toll worldwide. Perhaps, with a few exceptions, most of them were deceived in 2020 and did not understand that they were participating in a war crime and signing off on a deadly fraud. I believe most of them do know by now, I hope they are sufficiently horrified at what they have enabled, and I hope these people step forward as whistleblowers and start talking. We need answers.
Originally published by Due Diligence and Art
Suggest a correction