

Cosa rivelano le e-mail e i documenti trapelati dall’EMA: grandi preoccupazioni con l’integrità del lotto del vaccino Pfizer C-19 e la corsa per l’autorizzazione
Il nostro recente articolo di Meryl Nass, MD sul vaccino lanciato più velocemente nella storia del mondo, che descrive in dettaglio la procedura di approvazione per i nuovi “vaccini” mRNA, mette in evidenza l’incomprensibile ignoranza di come funzionano oggi le autorità sanitarie.
Allo stesso tempo, la commissione speciale del Parlamento europeo sulla pandemia di COVID-19 ha avuto l’opportunità di porre domande ai rappresentanti di quattro aziende farmaceutiche il 5 settembre 2022. L’incontro si è concentrato sulle attività legate alla pandemia del passato e del futuro con un tentativo disperato da pochi per portare la trasparenza e la responsabilità attese in un contesto democratico. Il Sig. Cristian Terhes, membro del Parlamento Europeo, ha sollevato le domande giuste.
Per tutti quelli che sono rimasti scioccati dalla rapida introduzione dei nuovi “vaccini” in Europa, dobbiamo ricordare e chiederci come sia andata l’autorizzazione della precedente politica di vaccinazione contro il COVID-19, come abbiamo appreso dalle e-mail trapelate dall’EMA (Agenzia europea per il farmaco), sintetizzato nel seguente articolo della giornalista investigativa ed emittente televisiva, Sonia Elijah, su Trial Site News (giugno 2022).
Trial Site News (video sopra) è stato recentemente in grado di rivedere le e-mail interne trapelate dall’Agenzia europea per i medicinali (EMA) e il rapporto dell’incontro tra l’agenzia e Pfizer. L’EMA sovrintende alla valutazione e alla supervisione dei medicinali per l’Unione Europea. Come altri organismi sanitari di regolamentazione, la sua principale responsabilità è proteggere e promuovere la salute pubblica. Istantanee della corrispondenza e-mail interna EMA; una presentazione PowerPoint del 26 novembre 2020 da un incontro fondamentale tra Pfizer e l’agenzia, nonché un rapporto Pfizer riservato di 43 pagine sono stati forniti da una fonte anonima a causa della loro fiducia nell’impegno di Trial Site per la trasparenza, l’accessibilità e la responsabilità in promozione di un’industria della ricerca biomedica altamente etica, incentrata sulla qualità e incentrata sulla salute pubblica.
Le agenzie di regolamentazione, come l’EMA, la Food and Drug Administration (FDA) negli Stati Uniti e la Medicines and Healthcare products Regulatory Agency (MHRA) del Regno Unito, sono istituite per prendere decisioni basate sul miglioramento del pubblico. Influenze esterne come la pressione politica o dei media non intendono essere un fattore trainante nel loro processo decisionale, tuttavia, quando si tratta di condizioni pandemiche e dell’autorizzazione all’immissione in commercio condizionale accelerata dei vaccini Covid-19 (in particolare per l’mRNA- a base di vaccini prodotti da Pfizer-BioNTech e Moderna), sembra che quest’ultima abbia vinto la gara.
Il periodo di tempo della corrispondenza e-mail in questione va dal 10 al 25 novembre 2020, poche settimane prima che l’EMA concedesse la CMA (autorizzazione all’immissione in commercio condizionale) per il vaccino Pfizer-BioNTech Covdid-19 il 21 dicembre 2020. La FDA ha concesso EUA (autorizzazione all’uso di emergenza) per questo vaccino l’11 dicembre con l’MHRA che è arrivato primo al traguardo il 2 dicembre. Qui questo autore usa il termine “traguardo”, poiché le e-mail rivelano un’intensa corsa quasi competitiva per autorizzare i vaccini Covid-19, il più rapidamente possibile. Comprensibilmente, all’epoca il mondo era attanagliato da una pandemia, in cui c’era un immenso impulso per autorizzare un vaccino per proteggere le persone dal nuovo coronavirus.
La corsa nell’UEA
In una e-mail, Marco Cavaleri, all’epoca Head of Biological Health Threats and Vaccines Strategy dell’EMA, comunicava con urgenza come la FDA statunitense “si precipiterà nell’EUA”.
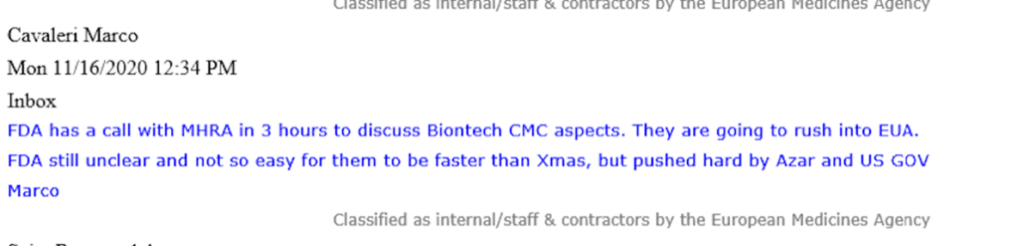
Cavaleri si riferisce a questa “corsa” che è stata “spinta duramente da Azar e dal governo statunitense”. Sotto l’amministrazione Trump, Alex Azar, l’ex dirigente farmaceutico è stato Segretario della salute e dei servizi umani (HHS) degli Stati Uniti dal 2018 al 2021. La FDA è un’agenzia che ricade direttamente sotto l’HHS.
Vale la pena notare che quando Azar era l’ex presidente della Lilly USA LLC, una divisione di Eli Lilly, i prezzi dei farmaci salirono alle stelle sotto la sua guida. La società farmaceutica è stata anche coinvolta in un’azione legale collettiva durante il suo mandato in cui è stata accusata di sfruttare il sistema dei prezzi dei farmaci per aumentare i profitti del suo farmaco insulinico. Ovviamente, questo non significa necessariamente che questo dirigente sia stato in alcun modo complice, ma il tempismo è degno di nota.
L’e-mail di Cavaleri parla di come la politica (e il governo degli Stati Uniti) stessero guidando il processo normativo della FDA, assicurandosi che stesse andando a “velocità di curvatura” (‘warp speed’). E, naturalmente, su quella nota, l’operazione Warp Speed di Trump doveva garantire che tutti i record di sviluppo del vaccino sarebbero stati infranti. Le intenzioni erano senza dubbio buone visto lo scoppio della peggiore pandemia in un secolo.
Tuttavia, dall’altra parte dell’Atlantico in Europa la tensione dell’agenzia di regolamentazione è aumentata poiché la pressione per accelerare le scadenze ha reso l’aria e l’umore generale tesi: la pressione e l’ansia erano palpabili negli scambi di e-mail esaminati.
Sono emerse persone di elevata integrità e chiarezza per quanto riguarda i loro ruoli e impegni come amministratori della salute pubblica. Ad esempio, un individuo ha dimostrato palpabile preoccupazione per le tempistiche accelerate per garantire che avrebbero rispettato la “scadenza” per l’autorizzazione del vaccino a scapito di una valutazione solida. Era Noel Wathion, all’epoca vicedirettore esecutivo dell’EMA, ma che da allora è andato in pensione. Questo funzionario dell’EMA ha sottolineato in modo importante: “Stiamo accelerando il più possibile, ma dobbiamo anche assicurarci che la nostra valutazione scientifica sia il più solida possibile”. Non dimentichiamo la responsabilità/responsabilità allegata alla raccomandazione alla CE di concedere un CMA.’

Wathion presume che l’EUA della FDA (e dell’MHRA) sarebbe stato emesso prima che l’EMA concedesse il proprio CMA, il che si è rivelato corretto. Ciò che è interessante è la sua preoccupazione nell’affrontare la “limitazione dei danni” derivante dal probabile esito dell’EMA che si è classificato ultimo in questa corsa alla regolamentazione e il suo timore che ciò possa portare l’opinione pubblica e i media a rivoltarsi contro l’agenzia. La velocità ha apparentemente superato i problemi di qualità sulla base di un’attenta revisione di queste e-mail.
In un’e-mail del 19 novembre, Wathion rivela un TC (teleconferenza piuttosto teso) con il commissario europeo (Ursula von der Leyen) che è stato “a volte anche un po’ spiacevole”. Ciò riflette la crescente pressione a cui era sottoposto il personale dell’EMA rilasciare rapidamente CMA a seguito di un EUA concesso dalla FDA/MHRA per il vaccino Pfizer-BioNTech. Von der Leyen è implicata nella potenziale responsabilità di questo ambiente teso con “un ritardo di diverse settimane… non facilmente accettabile per la CE [Commissione europea]”.
All’inizio del 2022, Trial Sites News ha riferito di come von der Leyen sia stata coinvolta in uno scandalo quando un gruppo di eurodeputati indipendenti ha chiesto le sue dimissioni immediate e la piena divulgazione di una serie di messaggi di testo privati tra lei e il CEO di Pfizer, Albert Bourla. Solo una piccola parte di questi testi è stata divulgata. Di quelli che lo erano, hanno rivelato che stava negoziando parti di un accordo sul vaccino a livello europeo, unilateralmente con Bourla tramite una serie di messaggi! Chiaramente i protocolli standard in Europa sono stati buttati fuori dalla finestra a favore dell’opportunità e questo apparentemente era legato a una pressione competitiva unificata su tutte e tre le agenzie di regolamentazione.
Wathion mette a nudo le sue riflessioni dopo questo particolare TC e scrive in modo sconvolgente come “le ricadute politiche sembrano essere troppo elevate anche se il livello “tecnico” degli Stati membri [Stati membri] potrebbe difendere un tale ritardo per ottenere il risultato della revisione scientifica più solida possibile.» In altre parole, la trasmissione continua della scienza è apparsa per la prima volta come copertura per la politica.

Wathion sottolinea che un potenziale ritardo di diverse settimane per garantire “una solida garanzia in particolare per quanto riguarda CMC e sicurezza” sarà accolto con “critiche da varie parti”, inclusi i media, la Commissione europea e il Parlamento europeo (Parlamento europeo). Wathion parla della sua paura che se la scadenza “per allinearsi il più possibile con i tempi di “approvazione” da parte della FDA/MHRA” non può essere rispettata, “saremo sopraffatti da tutti i fronti e saremo nel mezzo della tempesta”; questo potenziale ritardo sembrava necessario “per rendere il risultato della revisione scientifica il più solido possibile”. Ciò implica che la velocità a scapito della sicurezza era all’ordine del giorno per evitare “ricadute politiche”. Chiaramente, la politica stava dettando il protocollo di autorizzazione del vaccino Covid-19, non la scienza.

Nell’e-mail di cui sopra di Marco, il funzionario dell’EMA rivela che il CEO di Pfizer Albert Bourla ha “fatto pressioni” su Peter Marks, e questo potrebbe essere interpretato come molto controverso, dato che Marks è il direttore del Center for Biologics Evaluation and Research (CBER) presso la FDA . L’apparente accesso di Pfizer al watchdog federale solleva almeno questioni significative, se non introduce la possibilità di inquietanti intrecci tra l’industria e un’agenzia federale scientifica presumibilmente indipendente.
Principali preoccupazioni per l’integrità tra i lotti di vaccino
Un’e-mail di Cavaleri (vedi sotto) rivela che a quel tempo la FDA era a conoscenza di “alcuni problemi” associati alla CMC che dovevano essere risolti e avrebbero potuto “finire per essere la parte difficile”. CMC si riferisce alla chimica, produzione e controlli, denominata anche qualità farmaceutica, che copre varie procedure utilizzate per valutare e garantire la sicurezza e la coerenza tra i lotti di prodotti farmaceutici.
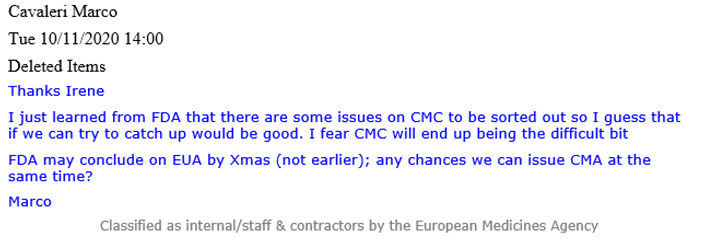
Un’e-mail di Evdokia Korakianiti (un amministratore scientifico dell’EMA) spiega in modo più dettagliato quali fossero questi “problemi” e come fossero in realtà le principali preoccupazioni a che fare con il vaccino Pfizer-BioNTech.

In modo allarmante, sono state osservate differenze significative nei livelli di integrità dell’mRNA tra i lotti di vaccini commerciali (su larga scala) e clinici (su piccola scala) di Pfizer-BioNTech. “~78% di integrità dell’mRNA” in quelli clinici e “~ 55% nei lotti commerciali proposti” con la “causa principale” non ancora identificata. Nell’e-mail sono state annotate anche le implicazioni di sicurezza ed efficacia dovute a questa preoccupazione “ancora da definire”.
In un rapporto riservato di Pfizer, trapelato anche insieme alle e-mail dell’EMA, la società afferma che secondo Acuitas Therapeutics’ (l’azienda biotecnologica che ha sviluppato la piattaforma di nanoparticelle lipidiche per il vaccino Pfizer e Moderna) possiede un’esperienza generale, “una soglia minima è di circa il 70%.’ (Vedi screenshot sotto)

Poi a pagina 30 si legge: L’efficacia del prodotto dipende dall’espressione dell’RNA somministrato, che richiede una molecola di RNA sufficientemente intatta“. (Vedi screenshot sotto)

Questa frase esatta “richiede una molecola di RNA sufficientemente intatta” è stata utilizzata nell’e-mail del personale dell’EMA, Evdokia Korakianiti, che ho incluso sopra, inviata il 23 novembre 2020 – ora probabilmente sappiamo da dove Korakianiti l’ha fatto riferimento.
Per i lotti commerciali (che sarebbero stati lanciati in tutto il mondo) avere un livello di integrità dell’mRNA significativamente inferiore (molecola di RNA intatta) è molto preoccupante, dato il suo legame intrinseco con l’efficacia e la potenziale sicurezza del prodotto.
Il giorno successivo Veronika Jekerle, responsabile dell’ufficio qualità della farmacia, scrive a Evdokia (vedi sotto).

La differenza nel livello di integrità dell’mRNA è stata nuovamente notata come una delle principali preoccupazioni “condivise dalla maggior parte degli Stati membri” e il suo “potenziale impatto sulla sicurezza”. Jekerle evidenzia in grassetto: “Potrebbe essere possibile un’approvazione entro la fine dell’anno, se queste preoccupazioni + GMP verranno risolte.
Ciò fa sorgere la domanda critica: come sono state risolte tutte queste preoccupazioni quando la CMA è stata concessa solo poche settimane dopo, il 21 dicembre? Un possibile modo in cui è stato risolto è spiegato più avanti in questo rapporto.
Contrariamente alle preoccupazioni di alcuni degli altri funzionari dell’EMA, Marco Cavaleri scrive più o meno nello stesso periodo nella seguente e-mail (vedi sotto) che il contenuto dell’mRNA non è una preoccupazione importante, secondo la FDA: “la questione sul contenuto dell’mRNA non percepito come importante.” Afferma anche in modo scioccante, “non è chiaro se le ispezioni GCP siano mai state eseguite”. Questa rivelazione è molto preoccupante dato che GCP si riferisce alla buona pratica clinica, che è “uno standard di qualità etico e scientifico internazionale per la progettazione, la conduzione, la registrazione e la segnalazione di prove che coinvolgono la partecipazione di soggetti umani.’
Ciò che è ancora più allarmante è la sua seguente dichiarazione: “nessun grande interesse da parte della FDA”.
Questo sembra rivelare l’apparente mancanza di preoccupazione o addirittura interesse dell’agenzia di regolamentazione sul completamento delle ispezioni GCP, nel contesto degli studi clinici di Pfizer, su cui si faceva affidamento dalla FDA per concedere EUA per il vaccino Pfizer-BioNTech. In uno dei precedenti rapporti investigativi di questo autore per Trial Site News, abbiamo notato che la FDA ha ispezionato solo l’1% dei siti di prova di Pfizer.

Ulteriori informazioni sul damming vengono rivelate (vedi screenshot sotto) quando più agenzie di regolamentazione: Health Canada (HC), EMA, MHRA e FDA sono tutte a conoscenza del problema con l’integrità dell’mRNA%, ma FDA e Health Canada affermano infondate che “problemi di sicurezza associato. Sono più di una preoccupazione teorica.’

Health Canada sembra quindi contraddirsi perché in seguito è stato descritto come mostra particolare preoccupazione per una regione che riceve “tutto il materiale non ottimale”. Ovviamente, non voleva essere quella regione.
Incredibilmente, la fine dell’e-mail rivela che “il richiedente [Pfizer] ha condiviso con la FDA e noi [EMA]/MHRA solo oggi e il problema con le particelle visibili nel DP [prodotto farmacologico] sembra essere componenti di nanoparticelle lipidiche.)”
Questo è molto preoccupante a causa del fatto che questo problema significativo è stato reso noto alle tre agenzie di regolamentazione chiave il 25 novembre, solo poche settimane prima che l’EMA concedesse CMA e la FDA concedesse EUA per il vaccino Pfizer. In modo allarmante, sono trascorsi pochi giorni prima che l’MHRA concedesse l’autorizzazione nel Regno Unito il 2 dicembre 2020. L’ipotesi di Veronika che le “particelle visibili” potrebbero essere LNP (nanoparticelle lipidiche) è difficile da accettare dato che le nanoparticelle non sono visibili ad occhio nudo. Altre anomalie erano evidenti, ma probabilmente si trattava ancora di uno sforzo storico in termini di velocità di sviluppo del vaccino. Sembra chiaro, tuttavia è stato necessario un po’ più di tempo.
In che modo la percentuale di integrità dell’mRNA% è stata apparentemente risolta
La discrepanza tra i lotti sembra essere stata risolta quando viene menzionato che “gli ultimi lotti [ricevuti dalla FDA] indicano che la % di RNA intatto è tornata intorno al 70-75%.”
Tuttavia, in un rapporto trapelato di un incontro con Pfizer e l’EMA il 26 novembre 2020, un giorno dopo l’e-mail di Veronika, rivela in modo sconvolgente che la specifica di integrità dell’RNA è stata rivista fino a >=50% per la durata di conservazione del prodotto farmaceutico, significativamente inferiore rispetto alla soglia minima del 70% che Acuitas Therapeutics aveva stabilito e alla media del 78% dei lotti clinici. Era questo il modo dell’EMA (e potenzialmente della FDA/MHRA/HC) di “risolvere” la questione per garantire “un’approvazione entro la fine dell’anno“?

Si fa menzione di “incertezze sulla coerenza della qualità del prodotto e quindi incertezza per quanto riguarda la sicurezza del prodotto e l’efficacia del prodotto commerciale”. Tuttavia, è sconcertante come l’abbassamento della specifica di integrità dell’RNA possa porre rimedio a questa grande obiezione.
In un’altra diapositiva l’artefatto afferma: “Le specie di RNA troncate [accorciate] e modificate dovrebbero essere considerate impurità correlate al prodotto”. Ciò conferma che queste specie di mRNA accorciato che hanno abbassato il livello di integrità percentuale dell’mRNA sono state classificate come impurità. Un’altra preoccupazione allarmante derivante da queste impurità è segnalata “la possibilità che proteine tradotte diverse dalla proteina spike (S1 S2) risultanti da specie di mRNA troncate e/o modificate dovrebbe essere affrontata“. (Vedi screenshot sotto)

Le prove in questo rapporto confermano che organismi di regolamentazione come FDA, MHRA, EMA e Health Canada erano a conoscenza delle differenze nei lotti, per quanto riguarda l’integrità dell’mRNA% e, per questo motivo, l’effetto su “sicurezza ed efficacia” era sconosciuto. Il rapporto trapelato della riunione Pfizer/EMA solleva preoccupazioni materiali presupponendo che il problema sia stato risolto semplicemente abbassando la specifica di integrità dell’RNA. In altre parole, forse non è mai stato risolto.
Un sito Web particolare che ha attirato molta attenzione di recente, che parla della differenza tra i batch è howbadismybatch.com. È un database completo con analisi su “codici lotto e decessi associati, disabilità e malattie per i vaccini Covid 19. Inserendo un numero di lotto di uno qualsiasi dei vaccini Covid-19, indica la frequenza degli eventi avversi segnalati associati a quel lotto.
Ho parlato con Sasha Latypova, che conduce sperimentazioni cliniche da oltre 25 anni e possiede la sua azienda di biotecnologie, per chiedere la sua opinione di esperti sui documenti trapelati. Lei disse:
“La mancanza di integrità dell’mRNA e la presenza di frammenti non caratterizzati di RNA in lotti del prodotto Pfizer è stata identificata come una “Obiezione principale” – una bandiera rossa regolamentare formale, considerata un’impurità del prodotto e sarebbe stata un ostacolo in qualsiasi normale processo di approvazione dei farmaci. Come minimo, è stato necessario un ulteriore studio clinico “ponte” per valutare gli effetti clinici che avrebbero richiesto mesi per essere progettati e condotti correttamente. Il panico ha annullato l’integrità scientifica ed è stato adottato uno standard di accettazione dei lotti arbitrariamente abbassato per rispettare una scadenza motivata politicamente. Ad oggi, questo problema rimane irrisolto e potrebbe essere la causa alla base dell’enorme variazione nei tassi di eventi avversi e decessi osservati per diversi numeri di lotto di produzione nel CDC VAERS e in altri database. “
Latypova ha fatto un riferimento appropriato al destino del Titanic, tracciando un confronto nel modo in cui gli organismi di regolamentazione hanno condotto il loro processo di “velocità di curvatura” per autorizzare i vaccini Covid-19. Il capitano del Titanic, Edward J. Smith, mirava a migliorare il tempo di traversata di un’altra nave, il che significava che la nave stava viaggiando troppo velocemente, in acque note per essere ghiacciate. Questo lo ha portato a una collisione fatale con un iceberg e il resto è storia.
Alla luce delle prove incluse in questo rapporto e del fatto che il vaccino Pfizer-BioNTech Covid-19 è uno dei prodotti più redditizi della storia (l’anno scorso Pfizer ha realizzato vendite per 37 miliardi di dollari con previsioni per il 2022 di 32 miliardi di dollari), questo autore si sforza di aprire una discussione con alcune questioni vitali che devono essere affrontate dalle agenzie di regolamentazione coinvolte, Pfizer e quelle della comunità scientifica/medica:
Quali sono le implicazioni di sicurezza ed efficacia di un’integrità dell’mRNA significativamente ridotta (derivante da mRNA troncato e modificato) nei lotti commerciali di questo vaccino?
Quali sono esattamente le particelle visibili osservate nel DP (prodotto farmacologico) che Pfizer ha condiviso all’ultimo minuto con EMA, FDA e MHRA e quali sono le sue implicazioni di sicurezza ed efficacia?
Le risposte a queste domande sono di grande importanza pubblica.
Trial Site News è stato recentemente in grado di esaminare le e-mail interne trapelate dall’Agenzia europea per i medicinali (EMA) e il rapporto dell’incontro tra l’agenzia e Pfizer. L’EMA sovrintende alla valutazione e alla supervisione dei medicinali per l’Unione Europea. Come altri organismi sanitari di regolamentazione, la sua principale responsabilità è proteggere e promuovere la salute pubblica. Istantanee della corrispondenza e-mail interna EMA; una presentazione PowerPoint del 26 novembre 2020 da un incontro fondamentale tra Pfizer e l’agenzia, nonché un rapporto Pfizer riservato di 43 pagine sono stati forniti da una fonte anonima a causa della loro fiducia nell’impegno di Trial Site per la trasparenza, l’accessibilità e la responsabilità in promozione di un’industria della ricerca biomedica altamente etica, incentrata sulla qualità e incentrata sulla salute pubblica.
Le agenzie di regolamentazione, come l’EMA, la Food and Drug Administration (FDA) negli Stati Uniti e la Medicines and Healthcare products Regulatory Agency (MHRA) del Regno Unito, sono istituite per prendere decisioni basate sul miglioramento del pubblico. Influenze esterne come la pressione politica o dei media non intendono essere un fattore trainante nel loro processo decisionale, tuttavia, quando si tratta di condizioni pandemiche e dell’autorizzazione all’immissione in commercio condizionale accelerata dei vaccini Covid-19 (in particolare per l’mRNA- a base di vaccini prodotti da Pfizer-BioNTech e Moderna), sembra che quest’ultima abbia vinto la gara.
Originariamente pubblicato su Trial Site News.
Si veda anche: Il mio rapporto trapelato sulle e-mail dell’EMA presentato al dottor Reiner Fuellmich e al suo Comitato Investigativo Corona su Sonia Elijah Substack qui.






