Lettere dal mondo sotterraneo
Credito immagine: Shutter Stock
Rivisitazione delle e-mail trapelate dall’EMA
Alla fine del 2020 una raccolta di documenti – circa 900 pagine di Pfizer dalla sezione “Vaccine” Chemistry Manufacturing and Controls (CMC) [“Vaccino” Chimica Manifattura e Controlli] della presentazione normativa all’Agenzia Europea per i Medicinali (EMA) – è trapelata ed è stata inviata a un numero di giornalisti. I documenti includevano anche scambi di e-mail da parte di alcuni revisori e alti dirigenti dell’EMA. Della fuga di notizie si è occupato il British Medical Journal, che ha potuto confermare l’autenticità dei documenti. Ho ricevuto questi documenti da un collega circa un anno dopo, alla fine del 2021, e ne ho letti e utilizzati molti nelle mie analisi. Nel mio lavoro professionale ho visto molti documenti relativi alla R&S di Pfizer, quindi posso anche confermare che questi documenti trapelati erano molto coerenti con la documentazione tipica di Pfizer. L’EMA non ne ha negato l’autenticità, limitandosi a dichiarare che le intestazioni di alcune e-mail erano state modificate.
Ora ho un numero molto maggiore di follower e una comprensione molto più chiara dell’organizzazione e della struttura pseudo-legale del cartello criminale che guida l’atrocità globale colloquialmente nota come “risposta alla pandemia di Covid”. Sto rivedendo le e-mail trapelate perché credo che forniscano alcune prove altamente significative.
I file di posta elettronica dell’EMA che ho letto contengono 14 screenshot di e-mail risalenti alla metà e alla fine di novembre 2020. Gli scambi provengono dal personale dell’EMA e da dirigenti di alto livello. A mio parere, queste e-mail dimostrano che:
- I revisori dell’EMA erano sottoposti a una massiccia pressione politica per inventare nuovi modi di approvare quei prodotti pericolosi e inapprovabili. Le pressioni provenivano dai vertici dei governi di Stati Uniti, Regno Unito e Unione Europea.
- Il commissario europeo Ursula von der Leyen ha fatto agli Stati membri promesse che non ha mai avuto intenzione di mantenere, al fine di legarli tutti in un unico patto per i contratti sui vaccini, prevenendo così qualsiasi decisione indipendente nei rispettivi Paesi.
- C’erano problemi gravi e irrisolvibili – data la tempistica volutamente irrealistica – con la qualità del prodotto che il personale dell’EMA è stato costretto ad accettare. Alcuni erano a disagio nel farlo e nell’esprimere le loro preoccupazioni. Altri hanno “sottovalutato” dati chiaramente inventati.
In definitiva, la revisione normativa in sé e le preoccupazioni sollevate non avevano importanza: il prodotto sarebbe stato commercializzato comunque. Ora sappiamo esattamente perché: le autorità di regolamentazione non avevano potere di regolamentazione su di esso. Le autorità di regolamentazione farmaceutica non supervisionano i materiali militari noti come “contromisure” e “dimostrazioni di produzione” (un linguaggio ambiguo che maschera gli agenti di guerra biologica prodotti dal governo statunitense acquisito e dai suoi partner globali). Le e-mail dimostrano che la maggior parte del personale dell’EMA erano attori inconsapevoli in questo gioco.
La conferma di ciò per il Regno Unito è emersa di recente.
Sulla base della risposta alla MHRA FOIA:
“Tutte le decisioni relative ai vaccini e alle autorizzazioni terapeutiche Covid sono state prese dal Ministro delle Licenze e non sono state delegate”
Traduzione – normalmente l’autorità di rivedere e approvare nuovi prodotti farmaceutici è formalmente delegata all’MHRA dal Segretario di Stato per la Salute (Regno Unito). Nel caso dei prodotti covid, la delega di autorità non esiste. Sembra che tutti questi provvedimenti siano stati adottati da Matt Hancock da solo (anche se lui punta il dito contro qualcuno più in alto). La stessa cosa è accaduta negli Stati Uniti: Alex Azar, sotto l’amministrazione Trump, ha impiegato sugli americani questi biomateriali non conformi, e Xavier Becerra, sotto Biden, continua a farlo oggi.
La pressione politica.
Il 16 novembre 2020 si è verificato il seguente scambio di e-mail tra alti dirigenti dell’EMA:
Noel Wathion – vicedirettore esecutivo (in pensione dal giugno 2021):

Agnes Saint-Raymond – Capo della Divisione Affari Internazionali:

Marco Cavaleri, presidente della Task Force Pandemia Covid-19 dell’EMA:

Le e-mail vanno lette dal basso verso l’alto.

Un paio di cose interessanti: le tre autorità di regolamentazione – FDA statunitense, MHRA britannica ed EMA dell’UE – sono tutte impegnate a coordinare i tempi di approvazione prima di qualsiasi revisione formale dei dati, prima che i comitati consultivi abbiano visto i risultati degli studi clinici, li abbiano discussi, li abbiano votati, ecc. Stanno discutendo le tempistiche perché i dati NON HANNO ALCUNA IMPORTANZA riguardo alla commercializzazione o meno di questi prodotti. Inoltre, stanno interagendo come se non fossero tre agenzie separate di nazioni sovrane distinte, responsabili di fronte a gruppi separati di contribuenti e alla supervisione del rispettivo Congresso/Parlamento, ma semplicemente dipartimenti burocratici già fusi in un unico governo globale. Infine, la FDA sta per “entrare di corsa nell’EUA”, “spinta da Azar” (Alex Azar – all’epoca segretario dell’HHS) e “Trump sta tirando le fila”.
Molte persone mi chiedono come sia possibile che migliaia di persone abbiano partecipato alla truffa orchestrata come “risposta alla pandemia di covid” – sicuramente non è possibile avere così tante persone in combutta! Non era necessario avere così tante persone al corrente. In questo caso, Noel Wathion, un alto dirigente dell’EMA, o non sa che la revisione dei dati è irrilevante ai fini dell’immissione sul mercato delle iniezioni, o la sta abilmente travisando (credo infatti che non ne fosse a conoscenza). Pertanto, non c’era bisogno che il personale dell’EMA al di sotto di lui ne fosse consapevole, e semplicemente bastava mettere loro fretta per svolgere qualsiasi compito ristretto sia stato loro assegnato. La compartimentalizzazione è la chiave per coprire qualsiasi truffa importante all’interno di grandi organizzazioni e strutture complesse. È questo il motivo per cui si è dimesso/ritirato poco dopo il lancio delle iniezioni mortali? È anche sotto pressione da parte della CE (Commissione Europea) per ottenere l’approvazione. E la Pfizer ora vuole un’autorizzazione all’immissione in commercio completa (MA) invece di quella condizionata (CMA)! Nota: la CMA è stata rilasciata, ma le condizioni non sono mai state soddisfatte da Pfizer/BioNTech, perché chi se ne frega, era un gioco fin dall’inizio.
“(Co)-Rapps” = correlatori. L’EMA è un’entità europea, composta dalle “autorità competenti” degli Stati membri, un tempo separate, che regolamentavano e approvavano i farmaci in ciascun Paese separatamente. Nella struttura europea, il gruppo di revisione tecnica e di co-revisione viene selezionato per un prodotto specifico. Nel caso dei “vaccini” covid, il team svedese guidato da Philip Josephson è stato il relatore (revisore principale) e il team francese guidato da Jean-Michel Race – il correlatore. “CHMP” = Comitato per i prodotti medicinali per uso umano (presso l’EMA).

*Cara Olga, ovviamente possiamo discutere lunedì su come fornire al meglio aggiornamenti alla CE sugli sviluppi in tempo reale di questi primi vaccini. Vediamo come ottenere questo risultato al meglio. Tre commenti che vorrei fare in aggiunta:
- La probabilità che la FDA (e anche l’MHRA) emetta un EUA prima che venga concesso una CMA è estremamente alta. Quindi dobbiamo prepararci per questo. Certamente il pubblico profano e i media non capiranno la sfumatura… per loro un’ “autorizzazione” è un’autorizzazione. Abbiamo opzioni per affrontare questo andando dalla limitazione dei danni alla gestione proattiva delle aspettative. Dobbiamo scegliere quale opzione è la migliore tenendo conto delle circostanze esatte.
- Stiamo accelerando il più possibile, ma dobbiamo anche assicurarci che la nostra valutazione scientifica sia la più solida possibile. Non dimentichiamo la responsabilità / obbligo di rispondere collegati alla raccomandazione alla CE di concedere una CMA. E abbiamo bisogno del supporto dei (Co)relatori e del CHMP per ottenere questo. Senza di loro non succederà.
- Il fatto che la compagnia adesso all’improvviso voglia ottenere una MA completa invece di una CMA può rendere le cose ancora più difficili…
Cordiali saluti, Noel*
L’e-mail è indirizzata a Olga Solomon della Commissione Europea, con in copia il capo di Noel, Emer Cooke – Direttore esecutivo dell’EMA ed ex dirigente dell’OMS. Questa è Emer Cooke:

Il patto intelligente di Ursula.
Ve la ricordate?

Ursula von der Leyen – Commissario UE, i cui successi includono la negoziazione di assurdi contratti di fornitura predatori di Pfizer per conto di tutti gli Stati membri dell’UE tramite sms con Albert Bourla, il CEO di Pfizer. In questi contratti i Paesi dell’UE dovevano mettere a garanzia beni statali, rinunciare a tutte le leggi sul controllo di qualità, sull’importazione e sulla protezione dei consumatori e rinunciare alla sovranità nazionale – per esempio, non potevano modificare la legislazione sulla responsabilità dei vaccini da parte dei propri parlamenti? I contratti predatori, che sono stati completamente censurati per proteggere i cosiddetti “interessi commerciali di Pfizer”. Il seguente scambio di e-mail si riferisce ai valorosi sforzi di Ursula:

*Cari Olga e Florian, solo un avviso: abbiamo appena terminato il TC tra il Commissario e l’ECDC/EMA in cui il Commissario ha posto domande sulla prevista approvazione del vaccino Pfizer e sui tempi dell’approvazione della FDA rispetto all’UE. Quindi, automaticamente è emersa la questione dell’Art 5(2) nazionale contro la CMA, che Noel ha spiegato in dettaglio e anche come intendiamo discuterne ulteriormente con le NCA la prossima settimana e in HMA. Guido ha anche suggerito di considerare di parlarne con i ministri della Salute dell’EPSCO. Sandra e Giorgios hanno detto che avrebbero discusso ulteriormente anche all’interno di SANTE, quindi, da qui la mia email a voi. (Era presente anche Andrzej). Da notare anche: il Commissario ha affermato che, dal momento che la CE si è impegnata con gli Stati membri e il Parlamento europeo, che i vaccini saranno disponibili per tutti gli Stati membri contemporaneamente – e che pertanto sarà importante che gli Stati membri non siano “costretti” a utilizzare tale rotta nazionale a causa di “ritardi” nella procedura formale di approvazione. Lei ha anche detto che è disposta a convocare personalmente i Ministri della Sanità competenti per evitare il ricorso all’Art 5(2). Cordiali saluti, Hilde*
Ci sono un mucchio di acronimi usati, i più rilevanti sono “EC” = Commissione Europea, “MS” = Stati Membri, “EP” = Parlamento Europeo. La frase chiave è che Ursula è “pronta a convocare personalmente i Ministri della Sanità competenti per evitare il ricorso all’articolo 5, paragrafo 2”. Di cosa si tratta? L’art. 5(2) si riferisce all'”art. 5(2) della Direttiva 2001/83″ – l’autorizzazione all’uso di emergenza in uno Stato membro europeo, emanata da ciascuno Stato membro separatamente nei propri Paesi. La CMA è un’autorizzazione condizionata all’immissione in commercio che viene rilasciata dall’EMA contemporaneamente a tutti i membri dell’UE. Pubblicizzata dall’UE come un processo molto più robusto di un’EUA (grassetto aggiunto da me):
…la CMA segue un quadro controllato e robusto che fornisce salvaguardie che le autorizzazioni per uso di emergenza potrebbero non avere. In realtà, un’autorizzazione all’uso di emergenza non è un’autorizzazione del vaccino, ma un’autorizzazione all’uso temporaneo del vaccino non autorizzato. La CMA garantisce che tutti i controlli di farmacovigilanza, i controlli di produzione, compresi i controlli dei lotti per i vaccini, e altri obblighi post-approvazione siano applicati in modo giuridicamente vincolante […]. In particolare:
-garantisce un monitoraggio rigoroso, attraverso il sistema di farmacovigilanza dell’UE, della sicurezza del farmaco in tutta l’UE. […]
-assicura il monitoraggio della sicurezza dopo l’autorizzazione e consente la raccolta di dati aggiuntivi in modo strutturato. […].
–La produzione rigorosa, compreso il rilascio dei lotti per i vaccini e la distribuzione, è soggetta agli stessi controlli continui di tutti i medicinali autorizzati. Il monitoraggio dei processi di produzione garantisce che il farmaco sia prodotto e controllato secondo elevati standard farmaceutici nel contesto di una commercializzazione su larga scala.
Nell’ambito di un’Autorizzazione all’Immissione in Commercio Condizionata (CMA) dell’UE, la responsabilità è del titolare dell’autorizzazione all’immissione in commercio. Il titolare dell’autorizzazione all’immissione in commercio sarà responsabile del prodotto e del suo uso sicuro.
In teoria sembra una cosa fantastica. Questo è ciò che Ursula ha promesso quando ha personalmente chiamato e forzato la mano ai politici degli Stati membri. Forse non è stato nemmeno necessario forzare loro la mano, visto che erano sufficientemente terrorizzati dalla propaganda covid e aspettavano che i “vaccini” miracolosi li salvassero. Il problema è che Ursula non ha mai avuto intenzione di mantenere queste promesse e, in ogni caso, non è possibile produrre i “vaccini” a base di mRNA con la sicurezza, l’efficacia e la qualità di produzione richieste dai prodotti farmaceutici. Ciò che Ursula voleva veramente da questo processo era legare insieme tutti gli Stati membri europei in un patto tramite la promessa di una CMA “robusta”, in modo che non potessero avere un’autorità indipendente sulle iniezioni distribuite nei loro Paesi. Il percorso dell’articolo 5 avrebbe significato che ogni Stato membro avrebbe potuto autorizzare il prodotto, con il potere di revocare l’autorizzazione in caso di problemi. L’articolo 5 prevede anche un esonero di responsabilità per il produttore, tuttavia rende impossibile l’obbligo del prodotto. Con il percorso della CMA, nessuno degli Stati membri avrebbe potuto esercitare un processo decisionale indipendente, e quindi lei sarebbe stata in grado di costringerli tutti a sottoscrivere gli stessi contratti, folli e quasi completamente censurati, di Pfizer, Moderna e AstraZeneca, che rinunciavano comunque a qualsiasi responsabilità e vietavano inoltre ai Paesi di modificare le proprie leggi in materia di responsabilità!
Gli acquirenti devono “indennizzare, difendere e tenere indenne Pfizer … da e contro qualsiasi causa, reclamo, azione, richiesta, perdita, danno, responsabilità, transazione, sanzione, multa, costo e spesa … derivanti da, relativi a, o risultanti dal Vaccino”
Obiezioni Maggiori notevoli e mancanza delle stesse da parte dei revisori dell’EMA.
La sezione “Chemistry Manufacturing and Controls” (CMC) della richiesta di licenza biologica è il pilastro principale dell’approvazione normativa. Descrive il processo di produzione e la conformità alle Buone Pratiche di Fabbricazione (cGMP) e ad un’ampia serie di leggi e regolamenti volti a garantire la purezza, la potenza, la consistenza e la sicurezza di farmaci e prodotti biologici fabbricati in serie. I dati sulla sicurezza e sull’efficacia ottenuti dagli studi clinici sono inutili se il produttore non è in grado di assicurare alle autorità di regolamentazione e alla comunità medica che: 1) il prodotto in questione, secondo le specifiche, è stato utilizzato negli studi clinici; 2) il prodotto è fabbricato in modo coerente, puro, di alta qualità, riproducibile, con un processo di produzione e fasi di controllo ben caratterizzati e prevedibili; 3) lo stesso prodotto sperimentato sarà distribuito in commercio.
Sono stati riscontrati problemi nella sezione CMC (Chimica, Manifattura e Controlli) della documentazione presentata da Pfizer:

I valutatori CMC non erano soddisfatti della tempistica fornita per la valutazione, poiché violava tutte le tempistiche normali e anche quelle accelerate, con un ampio margine. La soluzione è stata quindi quella di “spingere” i valutatori. Questo ha raggiunto un unico obiettivo: forzare le persone che avrebbero potuto sollevare delle preoccupazioni fino all’orlo dello sfinimento, in modo che si arrendessero e si accodassero. Dopotutto, i vertici sapevano bene che la revisione normativa non aveva alcun significato e alcuna implicazione sulla falsa “approvazione”, che sarebbe avvenuta a prescindere da tutto. Nel Regno Unito, l’MHRA (Agenzia Normativa sui Prodotti Medici e Sanitari) ha già ammesso di non avere alcuna delega formale per la revisione e l’approvazione di queste iniezioni, e sono pronto a scommettere che nemmeno l’EMA aveva tale autorità.
I valutatori di basso livello della CMC non lo sapevano e stavano lavorando molto duramente e molto probabilmente in buona fede. A fine novembre, hanno sollevato oltre 140 obiezioni formali alla presentazione della CMC di Pfizer, che aveva ancora molti buchi enormi e informazioni mancanti. Come riferimento, 10-15 obiezioni regolamentari normalmente impediscono a una domanda farmaceutica di andare avanti finché non vengono risolte. Tre Obiezioni Maggiori, ovvero bandiere rosse formali, sono discusse in modo specifico nelle e-mail che seguono. Io e altri abbiamo scritto ampiamente sulla MO#2 (mancanza di integrità dell’mRNA). Ecco un’e-mail di uno dei revisori, Evdokia Korakianiti, e una risposta di Alexis Nolte che discutono il problema e l’impatto (completamente sconosciuto e potenzialmente molto preoccupante) sull’efficacia e la sicurezza del prodotto:

Il problema della degradazione dell’mRNA è stato discusso anche dagli specialisti della qualità CMC Ton van der Stappen, esperto biofarmaceutico senior presso il Medicines Evaluation Board (con sede nei Paesi Bassi) e specialista della qualità per l’EMA:

e Brian Dooley, altro specialista della qualità farmaceutica dell’EMA:

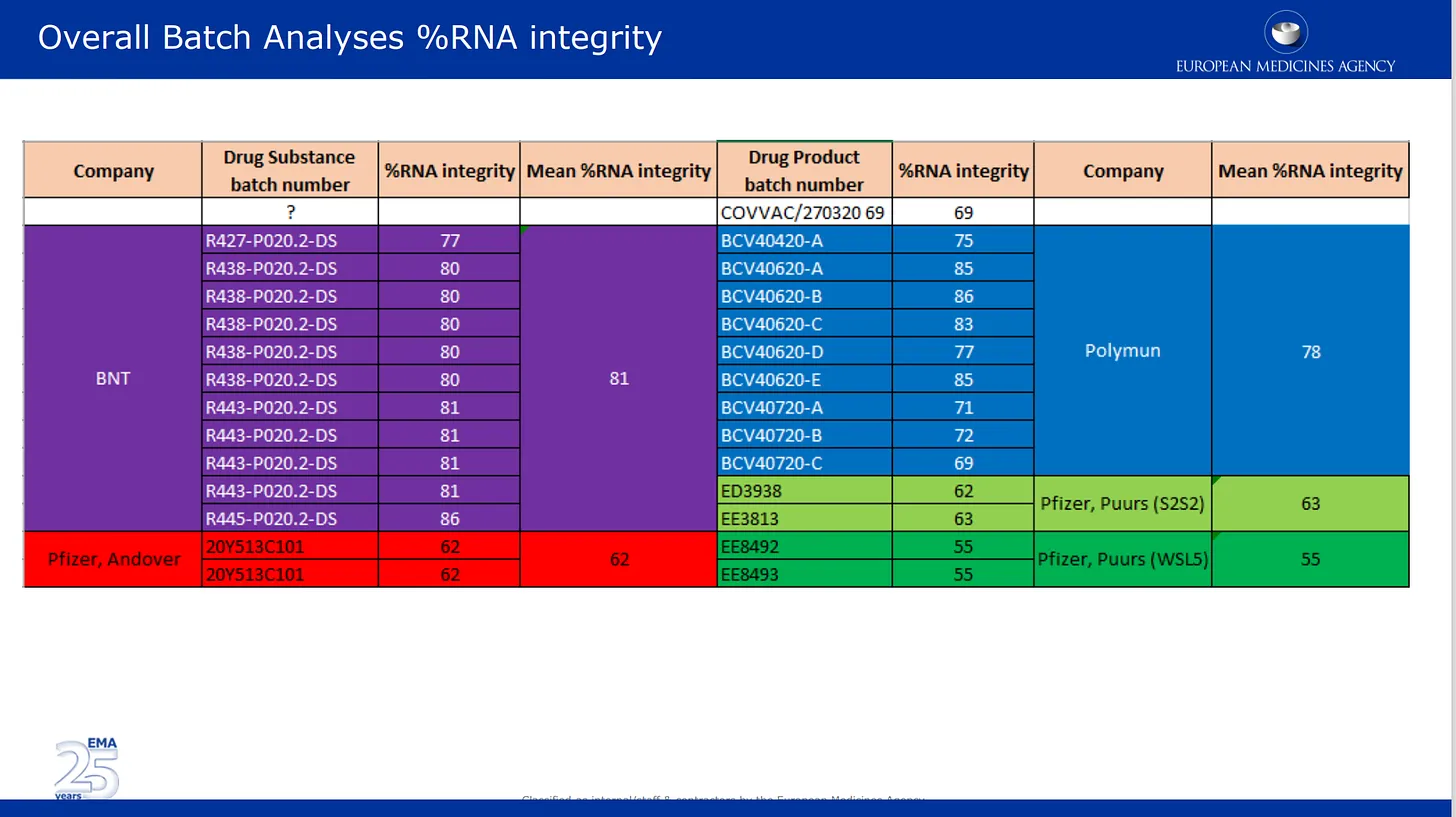
Le immagini che seguono sono tratte dalla revisione della qualità da loro presentata all’EMA. La prima immagine riguarda il problema, ormai ben documentato, della degradazione dell’mRNA in diversi lotti del prodotto Pfizer. Qui i risultati dell’analisi dei lotti forniti da Pfizer sono elencati e codificati per colori in base ai diversi stabilimenti di produzione, nonché in base alla sostanza farmaceutica e alla categoria del prodotto farmaceutico. Sostanza farmacologica = componenti attivi del prodotto (solo mRNA) e prodotto farmacologico è la sostanza formulata nei lipidi e negli altri ingredienti. La % di integrità dell’mRNA descrive la % di mRNA “a lunghezza intera” rilevata in un lotto. L’altra parte del lotto era composta da pezzi rotti sconosciuti con proprietà o impatto sulla sicurezza sconosciuti. Si noti che le autorità di regolamentazione non hanno effettuato alcun controllo indipendente, limitandosi a elencare i numeri forniti da Pfizer/BioNTech.
Sembra che questi due consulenti scientifici abbiano rivisto e accettato le false immagini dei risultati del Western blot presentate da Pfizer all’EMA – eccole nella loro presentazione PowerPoint di revisione del 24 novembre 2020. Leggete la nota sotto la diapositiva: stanno accettando queste immagini come reali, anche se entrambi i revisori dovrebbero saperlo bene. Perché non hanno sollevato obiezioni? Ecco cosa dice la nota:
La dimensione della proteina dopo l’espressione in vitro della sostanza farmacologica BNT162b2 è stata determinata mediante Western blot. La dimensione della proteina espressa è stata confermata come comparabile per i tre lotti del Processo 1 e per il lotto del Processo 2. La Figura 3.2.S.2.6-15 mostra che le dimensioni della proteina espressa sono coerenti con le dimensioni previste della sostanza farmacologica BNT162b2 e sono comparabili in tutti i lotti testati. Inoltre, i livelli di espressione relativa sono comparabili per tutti i lotti, come evidenziato dall’intensità della banda comparabile a ciascun livello di carico in tutti i lotti.

Forse qualche giornalista esperto dovrebbe contattare i dottori van der Stappen e Dooley, nonché la signora Korakianiti e le altre persone qui menzionate per un commento.
Dalle risposte fornite da Evdokia Korakianiti risulta evidente che il direttivo dell’EMA ha rinunciato alle armi e si è basato su “dati che solo la FDA ha visto”, ma è “ottimista” e che la FDA ha affermato che la rottura dell’mRNA era una “preoccupazione teorica”. Davvero? Ci sono dati a sostegno di questa affermazione o no? Ecco le e-mail che indicano che le Obiezioni Maggiori sono state formalmente scritte e successivamente ignorate dall’EMA, dal momento che il prodotto è stato spedito in commercio solo un paio di settimane dopo. Le condizioni della CMA non sono mai state soddisfatte.
Questo conferma ciò che già sappiamo: né l’EMA (né l’FDA, né Health Canada, né l’MHRA o altre autorità di regolamentazione) avevano alcuna autorità reale su questi prodotti o un impatto sul fatto che sarebbero stati distribuiti al pubblico ignaro. È stato tutto un teatro dall’inizio alla fine.
Ecco le tre principali obiezioni che rimangono tuttora irrisolte:

E qui c’è un po’ di agitare le mani e di accettare le affermazioni della FDA senza alcuna domanda o valutazione formale dei dati da parte dei regolatori dell’EMA:

Per concludere, posso dire che ho contato circa 70 persone diverse, citate nei documenti e nelle e-mail trapelate, che hanno facilitato questa tragica farsa: l’ “approvazione” del prodotto più letale che sia mai stato somministrato al maggior numero di persone, con un numero di morti e feriti senza precedenti in tutto il mondo. Forse, con poche eccezioni, la maggior parte di loro è stata ingannata nel 2020 e non ha capito che stava partecipando a un crimine di guerra e firmando una frode mortale. Credo che la maggior parte di loro ormai lo sappia, spero che siano sufficientemente inorriditi da ciò che hanno permesso, e spero che queste persone si facciano avanti come informatori e inizino a parlare. Abbiamo bisogno di risposte.
Pubblicato originariamente da Due Diligence and Art
Suggerisci una correzione