Eine weitere Untersuchung der durchgesickerten EMA-E-Mails und vertraulichen Pfizer-BioNTech-Dokumente zum Impfstoff COVID-19
Von Sonia Elijah
Im Juni veröffentlichte Trial Site News einen bahnbrechenden Untersuchungsbericht über die durchgesickerten E-Mails der Europäischen Arzneimittelagentur (EMA) und andere vertrauliche Berichte von Pfizer, in denen besorgniserregende Fakten im Vorfeld der Zulassung des Impfstoffs COVID-19 von Pfizer-BioNTech aufgedeckt wurden. Sie enthüllten:
- Ein politisch motivierter Wettlauf zwischen den wichtigsten Behörden bei der Zulassung des Impfstoffs.
- Ende November 2020 wussten die Aufsichtsbehörden, wie die US-amerikanische FDA, die Europäische Arzneimittelagentur (EMA), Health Canada und die britische MHRA, vom erheblichen Verlust der RNA-Integrität der kommerziellen Chargen (~55 % mRNA-Integrität) des Impfstoffs von Pfizer-BioNTech im Vergleich zu den klinischen Chargen (~78 % mRNA-Integrität). Dies wurde von der EMA zusammen mit den beobachteten sichtbaren Partikeln, die als „Verunreinigungen“ eingestuft wurden, als „wichtiger Einwand“ eingestuft.
- Eine am 26. November durchgesickerte PowerPoint-Präsentation eines Treffens zwischen Pfizer-BioNTech und der EMA enthüllte, wie dieser wichtige Einwand auf schockierende Weise „gelöst“ wurde – die Spezifikation für die RNA-Integrität wurde einfach auf 50 % gesenkt, d. h. die Hälfte aller mRNA-Moleküle in den kommerziellen Chargen durften trunkiert (nicht intakt) sein.
- Die möglichen Auswirkungen des Verlusts der RNA-Integrität auf die Sicherheit und Wirksamkeit waren nicht bekannt.
Dieser Bericht befasst sich mit weiteren durchgesickerten E-Mails, insbesondere mit denen von EU-Kommissarin Ursula von der Leyen und dem ungewöhnlichen Ausmaß, in dem sie bereit war, die Mitgliedstaaten dazu zu bringen, die Anwendung von Artikel 5 (2) (ihre nationale Notfallzulassung für die COVID-19-Impfstoffe) zu vermeiden und stattdessen eine EU-Zulassung für das bedingte Inverkehrbringen (Conditional Marketing Authorization) zu erteilen. Der Bericht beleuchtet weitere durchgesickerte sensible Dokumente der EMA: die Präsentation der CMC-Beobachtungen des Qualitätsbüros vom 24. November 2020 durch die BWP (Biologics Working Party) und den Bewertungsbericht des Berichterstatters für die turnusmäßige Überprüfung, der weitere Beweise für die im ursprünglichen Bericht von Trial Site News diskutierten „großen Einwände“ offenbart. Der Bericht enthält ungeschwärzte Versionen von Pfizer-Impfstoffverträgen und die im November 2020 mit Pfizer und BioNTech unterzeichnete Vorabkauf Vereinbarung (Advance Purchase Agreement) „für die Entwicklung, Produktion, vorrangige Kaufoptionen und Lieferung eines erfolgreichen COVID-19-Impfstoffs für die EU-Mitgliedstaaten.
Skandale sind dieser Frau keinesfalls fremd
Ursula von der Leyen stand unter intensiver öffentlicher Beobachtung, weil sie die Verhandlungen über einen milliardenschweren Impfstoffvertrag (den größten Vertrag der EU) über geheime Texte und Telefongespräche mit Albert Bourla, dem Vorstandsvorsitzenden von Pfizer, führte und damit gegen den Beschluss der Kommission verstieß, einen Lenkungsausschuss einzurichten, der „während des gesamten Bewertungsprozesses beratend tätig sein sollte“. Außerdem weigerte sie sich, der Aufforderung nachzukommen, der Öffentlichkeit Zugang zu den geheimen Textnachrichten zu gewähren, die zwischen Bourla und ihr selbst ausgetauscht wurden. Der Europäische Rechnungshof veröffentlichte einen alarmierenden Bericht, in dem es heißt: „Wir haben die Kommission gebeten, uns Informationen über die Vorverhandlungen zu diesem Abkommen zu übermitteln (wie über erhaltene Ratschläge von konsultierten wissenschaftlichen Experten, Gesprächsprotokolle, Gesprächsaufzeichnungen und Einzelheiten zu den vereinbarten Bedingungen). Es wurden jedoch keine Informationen vorgelegt.
Auch für Bourla von Pfizer wird es immer brenzliger, denn er sollte vor dem Sonderausschuss des Europäischen Parlaments zu COVID-19 offiziell aussagen, um sich den Fragen zu den geheimen Impfstoffgeschäften zu stellen, zog aber in letzter Minute seine Zusage sich dem Ausschuss zu stellen, zurück.
Die geleakten E-Mails
Der ursprüngliche Trial Site News-Untersuchungsbericht verdeutlichte den enormen Druck, den die Europäische Kommission auf die EMA ausübte, um die bedingte Zulassung innerhalb eines stark beschleunigten Zeitrahmens zu erteilen. Eine E-Mail von Noel Wathion (ehemaliger stellvertretender Exekutivdirektor der EMA) wurde veröffentlicht, die eine „ziemlich angespannte“ Telefonkonferenz mit der EU-Kommissarin (Ursula von der Leyen) enthüllte, die „zeitweise sogar etwas unangenehm“ war.
Eine Verzögerung von mehreren Wochen war für die EK [Europäische Kommission] nicht ohne weiteres akzeptabel“, so Wathion. Er zeigt auch auf, „dass die politischen Auswirkungen zu groß zu sein scheinen, selbst wenn die „technische“ Ebene in den Mitgliedstaaten eine solche „Verzögerung“ verteidigen könnte, um das Ergebnis der wissenschaftlichen Überprüfung so solide wie möglich zu gestalten“.
Nachfolgend finden Sie eine E-Mail von Hilde Boone von der EMA, in der sie [von der Leyen] bereit ist, die zuständigen Gesundheitsminister persönlich anzurufen, um die Anwendung von Artikel 5 (2) zu vermeiden“.

Von: Boone Hilde <Hilde boone@ema.europa.eu>
Gesendet: Donnerstag, November 12, 2020 3:57 PM
To: SOLOMON Olga (SANTE) <Olga.Solomon@ec.europa.eu~; SCHMIDT Florian (SANTE)“Florian.SCHMIDT@ec.europa.eu=Cc: GIRARD Thomas (EMA) „Thomas Girard@ema.europa.eu>; CAVALERI Marco (EMA <Marco.Cavaleri@ema.europa.eu-;WATHION Noel (EMA) <Noel.Wathion@ema.europa.eu~
Subject: Art 5(2) vs CMA
Liebe Olga & Florian
nur eine Vorwarnung: wir haben gerade das TC zwischen der Kommissarin und ECDC/EMA beendet, in dem die Kommissarin Fragen zur erwarteten Zulassung des Impfstoffs von Pfizer und zum Zeitplan zwischen FDA und EU-Zulassung. So kam automatisch die Frage des nationalen Art 5(2) vs. CMA auf, Noel erläuterte dies im Detail und erklärte auch, wie wir dies mit den nationalen Wettbewerbsbehörden nächste Woche und in der HMLA diskutieren wollen. Guido schlug auch vor, das Thema bei den Gesundheitsministern im EPSCO ANZUSPRECHEN.
Sandra und Giorgios sagten, sie würden das Thema auch innerhalb der SANTE weiter diskutieren, daher meine E-Mail an Sie.
(Andrzej war ebenfalls anwesend)
Ebenfalls erwähnenswert:
Die Kommissarin sagte, dass die EG gegenüber den Mitgliedstaaten und dem EP die Verpflichtung eingegangen ist, dass die Impfstoffe allen Mitgliedstaaten zur gleichen Zeit zur Verfügung stehen werden – und dass es daher wichtig ist, dass die Mitgliedstaaten nicht „gezwungen“ werden, diesen nationalen Weg zu gehen, um „Verzögerungen“ zu vermeiden durch ein formelles Genehmigungsverfahren.
Sie sagte auch, dass sie bereit sei, die zuständigen Gesundheitsminister persönlich anzurufen, um die Anwendung von Artikel 5(2) zu vermeiden.
Mit freundlichen Grüßen,
Hilde
Article 5 (2) vs CMA
Artikel 5 (2) gegen CMA
In Artikel 5 (2) der Richtlinie 2001/83 heißt heißt es: „Die Mitgliedstaaten können als Reaktion auf die vermutete oder bestätigte Ausbreitung von Krankheitserregern, Toxinen, chemischen Stoffen oder nuklearer Strahlung, die Schaden verursachen können, vorübergehend die Verteilung eines nicht zugelassenen Arzneimittels genehmigen. Mit anderen Worten, es handelt sich um das Äquivalent einer Notfallgenehmigung auf der Ebene der Mitgliedsstaaten. Sie kann sehr schnell erteilt werden, da das Arzneimittel nicht das übliche nationale Zulassungsverfahren durchlaufen muss.
Ein Experte für EU-Recht erklärte Trial Site News, dass der Nachteil der Anwendung von Art. 5 (2) nutzen, ist, dass dies zu einem Wettbewerb zwischen den Mitgliedstaaten geführt hätte, der einen ungleichen Zugang/Verteilung der COVID-19-Impfstoffe zur Folge gehabt hätte, insbesondere für diejenigen Mitgliedstaaten, die es vorzogen, auf die EU-Zulassungsbehörde zu warten (was länger dauert, da es einem kontrollierten und robusten Rahmen mit Schutzbestimmungen folgen soll). Eine EU-Zulassungsgenehmigung trug dazu bei, dass die Impfstoffe allen Mitgliedstaaten zur gleichen Zeit zur Verfügung gestanden hätten, was eines der Ziele der COVID-19-Strategie der EU war.
Könnte es jedoch noch weitere Gründe gegeben haben, warum von der Leyen unbedingt wollte, dass die Mitgliedstaaten Art. 5 (2) umgehen, so dass sie bereit war, jeden zuständigen Gesundheitsminister selbst anzurufen?
In der nachstehenden E-Mail von Noel Wathion heißt es: „Da die erste Option immer noch darin besteht, eine CMA anzustreben und nicht einen Artikel 5 (2), der mit einem Artikel 5 (3) gekoppelt ist oder nicht“…

Fra: Wathion Noel <Noel.Wathion@ema.europa.eu‘
Sendt: 20. november 2020 10:16
Til: Thomas Senderovitz<THS@dkma.dk>
Cc: Cooke Emer<Emer.Cooke@ema.europa.eu>
Emne: RE: von der Leyden Aussagen
Lieber Thomas,
ich habe Emer in meiner Antwort cc.
An sich hat sich nicht viel geändert, da sie klar sagt „wenn alles gut geht“. Und ja, das stimmt mit der EMA und den Aussagen überein, die wir in mehreren Interviews gemacht haben, seit die Ergebnisse der Zwischenanalyse für den Impfstoff von Pfizer/BioNTech (diese Woche durch die endgültigen Analyseergebnisse bestätigt) und die Ergebnisse der Interimsanalyse für den 2. m-RNA-Impfstoff für Moderna bekannt gegeben wurden.
Neu ist meines Erachtens, dass sie die beiden Impfstoffe, die noch vor Ende des Jahres zugelassen werden könnten, klar benennt. Bei beiden gibt es noch Probleme (CMC scheint ein Problem für Pfizer/BioNTech zu sein, und die fortlaufende Überprüfung für Moderna hat gerade begonnen, so dass weniger Zeit für die Überprüfung zur Verfügung steht), so dass man abwarten muss, ob all dies rechtzeitig geklärt werden kann, ohne dass die Qualität der Prüfung beeinträchtigt wird.
In Anbetracht der Tatsache, dass die rechtlichen Instrumente in den USA und in der EU unterschiedlich sind, und da die erste Option immer noch darin besteht, eine CMA anzustreben, anstatt ein Verfahren nach Artikel 5(2) in Verbindung mit oder ohne Art. 5(3) anzustreben, versuchen wir, das CMA-Verfahren so weit wie möglich an die Pandemie-Situation anzupassen, da es bisher hauptsächlich für Onkologie-Arzneimittel verwendet wurde.
Die Diskussion auf der EU Exe SG am Mittwoch war sehr nützlich, und wir werden diese Arbeit beschleunigen.
Damit komme ich zum nächsten Punkt, nämlich der Frage, wie wir die Angleichung innerhalb des Netzes am besten fortsetzen können; für die nächsten Wochen sind folgende Treffen geplant, um COVID-19 zu diskutieren:
- HMA am Donnerstag: 10 Minuten Update durch die EMA und ein spezielles Thema zu klinischen Versuchen, wenn ich mich nicht irre, das vom PEI vorgestellt wird.
- EU Exe SG in der übernächsten Woche, wo der Punkt, das CMA-Verfahren so effizient wie möglich zu gestalten, erneut auf der Tagesordnung steht.
- MB am 16-17/12.
Da wir es mit einer außergewöhnlichen Situation zu tun haben, die für das Netzwerk und seine Glaubwürdigkeit von größter Bedeutung ist, sollten wir es in den nächsten 6 Wochen bewältigen (wir befinden uns im Hotspot für die ersten Impfstoffe, der Druck dürfte geringer sein, bei diesem Wirkungsgrad und unter der Voraussetzung, dass es keine größeren Qualitäts- und Sicherheits-probleme gibt), also sollten wir nicht eine dringende Ad-hoc-Sitzung HMA, EMA, EG (für etwa 3 Stunden) organisieren, um eine eingehendere Diskussion über alle Engpässe zu führen zur Beschleunigung des CMA-Prozesses und uns auch über die Kommunikation für die verschiedenen Szenarien abstimmen (frühere Zulassung durch FDA/MHRA, ähnliche Zeitpläne, Abwägung zwischen Geschwindigkeit und Sicherheit)? Oder alternativ die HMA-Sitzung am nächsten Donnerstag oder die EU-Exe-SG-Sitzung in der darauf folgenden Woche verlängern? Aber ich würde nicht viel länger als bis Mittwoch nächster Woche warten.
Ich freue mich auf Ihr Feedback.
KR,
Noel
Article 5(3) states: ‘Member States shall lay down provisions in order to ensure that marketing authorisation holders, manufacturers and health professionals are not subject to civil or administrative liability for any consequences resulting from the use of a medicinal product…’
The fact that Wathion points out that an Art. 5 (2) can be coupled or not with an Art. 5 (3), raises the important question whether member states would have had the option to not give indemnity to the marketing authorisation holders (in this case BioNTech) and manufacturers (BioNTech and Pfizer).
The Predatory Pfizer Contracts and Advance Purchase Agreements
Given what we know about the leaked Pfizer contracts made available by the non-profit consumer advocacy organization, Public Citizen; this pharmaceutical company has seemingly bullied countries into silence; can go after sovereign state assets (under the waiver of sovereign immunity) and enjoy full indemnity- exemption from legal liability that may result from their product- in fact it’s the Purchaser who is made liable on their behalf. This means governments have had to pay compensation to citizens who have suffered from a vaccine adverse event, not the vaccine manufacturer.
The screenshot below is taken from the unredacted version of the advance purchase agreement (APA) between the European Commission (EC), acting on behalf and in the name of member states, Pfizer Inc. and BioNTech (collectively ‘the Contractor’) signed in November 2020 (around the same time the leaked EMA emails were generated). It states ‘each Participating Member State shall indemnify and hold harmless the Contractor, their Affiliates..from and against any and all liabilities incurred..relating to harm, damages and losses..arising from or relating to the use and deployment of the Vaccines..’

However, information posted on the EC’s website states that under an EU Conditional Marketing Authorisation (CMA), ‘liability is with the holder of the marketing authorisation’ (which in this case is BioNTech). This directly conflicts with the APA that the EC signed with Pfizer and BioNTech (for 200 million vaccine doses priced at 15.50 Euros per dose excl VAT), a month before CMA was granted. It’s noteworthy that the EC published a heavily redacted version of the identical APA, any section related to indemnity/liability or simply considered ‘sensitive’ has been censored.
Another important point to consider when it comes to Art 5 (2) vs an EU CMA- is that given some European countries mandated the vaccine for adults, at-risk age groups and certain job sectors – it’s highly unlikely those MSs would have been able to do that with an unauthorised medicinal product, which these vaccines would have been classified under Art 5 (2).
The CMC issues
Wathion’s November 20, 2020 email raises several concerning points -‘there are still issues with both (CMC seems to be a concern for the Pfizer/BioNTech and the rolling review for Moderna just started giving less time to review) so it needs to be seen if all this can be sorted out on time, whilst not compromising the robustness of the review.’
The original Trial Site News report discussed what those CMC (Chemistry Manufacturing and Controls) issues were with Pfizer/BioNTech, particularly the loss of RNA integrity in the commercial batches and the unknown visible particles observed. Wathion tellingly states, ‘there are still issues’ speaks to the notion these ‘issues’ were not solved but had been ongoing. Wathion’s concern of ‘compromising the robustness of the review’ because of the need to authorise ‘on time’ is emphasized. This worry of speed over safety is reflected in other leaked emails, particularly by Wathion.
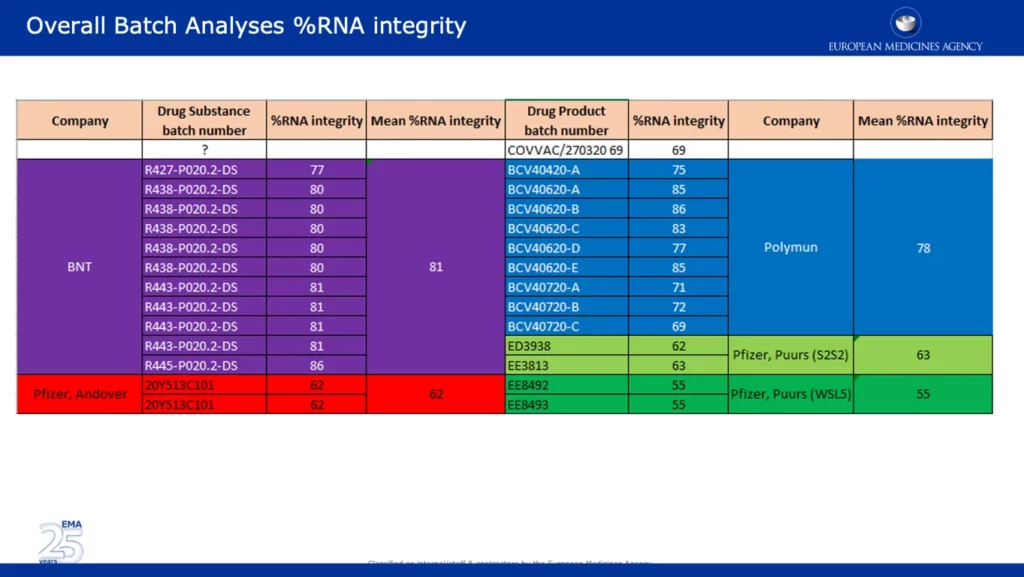
The report also contained a leaked email from Veronika Jekerle, EMA’s Head of Pharmaceutical Quality Office, where she outlined the 3 agreed major objections and the conclusion of the BWP (Biologics Working Party) regarding the Pfizer-BioNTech vaccine. Her email was sent on the November 24th the same day as the BWP presentation. Below, are a series of screenshots of the actual leaked BWP power point presentation (which Jekerle references in her email), entitled ‘EMA Quality Office CMC Observations.’ At the end of her email she thanks, ‘Ton, Brian and Claudio.’ Ton van der Stappen and Brian Dooley are named in the screenshot of the slide below.

The screenshot below presents wide-ranging data, which backups one of the major EMA objections in reference to the significant drop in %RNA integrity between the clinical and commercial batches. It’s noteworthy that batches from Pfizer’s Andover, USA (~62%) and Puurs, Belgium (~55%) sites were reported as having significantly lower %RNA integrity than batches provided by BNT (BioNTech) and Polymun.
The screenshot below is taken from the unredacted APA signed by the EC. It specifies that the majority of Europe’s vaccine supply will ‘come from Pfizer’s manufacturing site in Puurs, Belgium.’ This is concerning given the data shows that the %RNA integrity of batches from that site were the lowest at 55%, compared to other sites.

The screenshot below shows the acceptance criteria of various BNT 162B2 Drug Product lots. Drug Product refers to ‘medication in its marketed form, including its fillers, coloring agents, and other active or inactive agents.’ The acceptance criteria for Emergency Supply is greater or equal to 50%, which happens to be just below the batches with the lowest %RNA integrity supplied by Pfizer, Puurs. For some reason the acceptance criteria for the clinical Drug Product Lots differs and is set higher at greater or equal to 60%.

CMC (Chemistry, Manufacturing and Controls) ‘involves defining manufacturing practices and product specifications that must be followed and met in order to ensure product safety and consistency between batches.’ Given the data shown above, it’s evident there were significant CMC issues regarding the inconsistency of the %RNA integrity between batches, which is reflected in the leaked EMA emails and documents. What’s concerning is that the manufacturer (Pfizer/BioNTech) claimed, “The efficacy of the drug product is dependent on the expression of the delivered RNA, which requires a sufficiently intact RNA molecule.”
It’s baffling how lowering the specification down to 50% was potentially how this major objection was ’solved.‘ According to a leaked Rapporteur’s Rolling Review Assessment (revised report date: 25 November, 2020) compiled by the CHMP and PRAC rapporteurs- they found the ‘current acceptance criteria’ particularly troubling. The report states, ‘no characterisation data on RNA integrity and truncated forms is presented and the potential safety risks associated with truncated RNA isoforms are not addressed. This is especially important considering that the current DS and DP acceptance criteria allows for up to 50% fragmented species.’

Truncated (shortened by missing either top or end section) RNA is defined as a ‘product-related impurity’ and the fact that potential safety risks arising from this fragmented species ‘are not addressed’ is highly alarming.
Reference is made to ‘the current DS and DP acceptance criteria,’ this implies that it was not always set at that level and perhaps it had been changed (lowered). The question is why? Could it have been that process 2 (the manufacturing of the commercial batches) was just not replicable at the same specification level to the clinical batches (small scale) of process 1, therefore a lower standard was set in order for CMA to be granted?
Trial Site News communicated with the EMA regarding the content of the leaked emails and documents. The EMA press office’s prompt response has been published below in its entirety.
“The investigation of the published material revealed that the correspondence was manipulated by the perpetrators prior to publication. Not all of the documents were published in their integral, original form and may have been taken out of context. Whilst individual emails were authentic, data from different users were selected and aggregated, screenshots from multiple folders and mailboxes were created and additional titles were added by the perpetrators.
These documents do not present a full picture of the assessment of Comirnaty, the COVID-19 vaccine developed by BioNTech/Pfizer. They show the situation up to the beginning of December 2020, when the hack was discovered, but do not mention the considerable amount of additional data, information and clarifications submitted by BioNTech/Pfizer up to 21 December 2020, the day when EMA’s committee for human medicines (CHMP) gave its recommendation to grant a marketing authorization for this vaccine.
Comirnaty works because the mRNA it contains provides instructions for producing a spike protein which triggers an immune response. Its efficacy therefore depends on the presence of suitable amount of intact mRNA, which is known to be relatively unstable. What the documents show is how the assessment of any medicine works: following scrutiny of the data submitted by the company, the CHMP had questions about the integrity of mRNA and raised them formally as a ‘major objection’. This is an integral part of the assessment of any medicine. If major objections remain unresolved, they preclude the granting of the marketing authorisation. In this case, the company addressed the issues raised satisfactorily and subsequently supplied the required information and data after beginning of December 2020, which allowed EMA to move towards a positive opinion for this vaccine.
The public assessment report of Comirnaty summarises the conclusions of the CHMP on this issue and details the steps taken during the marketing authorization procedure of Comirnaty as well as the obligations placed on the marketing authorisation holder to carry out additional studies to closely monitor the pharmaceutical quality of vaccine. These obligations were also included in the Product Information published at the time of the CHMP opinion.
Even in a public health emergency as COVID-19, there has always been consensus across the EU not to compromise standards and to base any recommendation on the available scientific evidence on a vaccine’s safety, pharmaceutical quality and efficacy, and nothing else. Authorizations are only granted when the evidence shows convincingly that the benefits of vaccination are greater than any risks posed by a vaccine.”
Originally published in Trial Site News.
Korrektur vorschlagen