

Briefe aus der Unterwelt
Bildnachweis: Shutter Stock
Durchgesickerte EMA-E-Mails im Rückblick
Von Sasha Latypova
Ende 2020 wurde eine Sammlung von Dokumenten – etwa 900 Seiten – aus Pfizers „Impfstoff“-Abschnitt „Chemistry Manufacturing and Controls“ (CMC) des Zulassungsantrags bei der Europäischen Arzneimittel-Agentur (EMA) veröffentlicht und an eine Reihe von Journalisten weitergeleitet. Die Dokumente enthielten auch den E-Mail-Verkehr zwischen einigen der Prüfer und leitenden Angestellten der EMA. Über das Leck wurde im British Medical Journal berichtet, das bestätigen konnte, dass die Dokumente authentisch waren. Ich habe diese Dokumente etwa ein Jahr später, Ende 2021, von einem Kollegen erhalten und viele von ihnen gelesen und für meine Analysen verwendet. Ich habe im Rahmen meiner beruflichen Tätigkeit viele FuE-bezogene Dokumente von Pfizer gesehen und kann daher bestätigen, dass diese durchgesickerten Dokumente in hohem Maße mit typischen Pfizer-Dokumenten übereinstimmen. Die EMA hat die Authentizität nicht bestritten, sondern lediglich erklärt, dass die Kopfzeilen einiger E-Mails geändert wurden.
Ich habe jetzt eine viel größere Zahl von Anhängern und ein viel klareres Verständnis der Organisation und der pseudo-legalen Struktur des kriminellen Kartells, das die globale Gräueltat antreibt, die umgangssprachlich als „Covid-Pandemie-Bekämpfung“ bekannt ist. Ich schaue mir die durchgesickerten E-Mails noch einmal an, da ich glaube, dass sie einige sehr wichtige Beweise liefern.
Die E-Mail-Dateien der EMA, die ich gelesen habe, enthalten 14 Screenshots von E-Mails von Mitte bis Ende November 2020. Der Austausch stammt von Mitarbeitern und Führungskräften der EMA. Meiner Meinung nach zeigen diese E-Mails, dass:
- Die EMA-Prüfer standen unter massivem politischem Druck, neue Wege zu erfinden, um die nicht genehmigungsfähigen gefährlichen Produkte zu genehmigen. Der Druck kam von ganz oben aus den Regierungen der USA, Großbritanniens und der EU.
- EU-Kommissarin Ursula von der Leyen machte den Mitgliedstaaten Versprechungen, die sie nie einhalten wollte, um sie alle in einen einzigen Pakt für Impfstoffverträge einzubinden und so jede unabhängige Entscheidung in ihren eigenen Ländern zu verhindern.
- Es gab schwerwiegende und – angesichts des absichtlich unrealistischen Zeitplans – nicht zu lösende Probleme mit der Qualität des Produkts, zu dessen Genehmigung die EMA-Mitarbeiter gedrängt wurden. Einigen war es unangenehm, dies zu tun und ihre Bedenken zu äußern. Andere „übersahen“ eindeutig erfundene Daten.
Letztendlich spielten die behördliche Prüfung selbst und die geäußerten Bedenken keine Rolle – das Produkt wurde trotzdem auf den Markt gebracht. Wir wissen jetzt genau, warum – die Aufsichtsbehörden hatten keine Regelungsbefugnis für das Produkt. Die pharmazeutischen Aufsichtsbehörden beaufsichtigen keine militärischen Materialien, die als „Gegenmaßnahmen“ und „Herstellungsdemonstrationen“ bekannt sind (eine verdeckte Sprache, die die von der gefangenen US-Regierung und ihren globalen Partnern hergestellten Biokampfstoffe verschleiert). Die E-Mails zeigen, dass die meisten Mitarbeiter der EMA unwissentlich an diesem Spiel beteiligt waren.
Dies wurde kürzlich auch für das Vereinigte Königreich bestätigt.
Ausgehend von der Antwort auf die FOIA-Anfrage der MHRA:
„Alle Entscheidungen über die Zulassung von Covid-Impfstoffen und Therapeutika wurden vom Licensing Minister getroffen und wurden nicht delegiert.“
Übersetzt heißt das: Normalerweise wird die Befugnis zur Überprüfung und Zulassung neuer Arzneimittel vom britischen Gesundheitsminister formell an die MHRA delegiert. Im Falle der Kovid-Produkte gibt es diese Befugnisdelegation nicht. Es hat den Anschein, dass alle diese Maßnahmen von Matt Hancock im Alleingang ergriffen wurden (auch wenn er mit dem Finger auf jemand Höheren zeigt). Das Gleiche geschah in den USA – Alex Azar unter der Trump-Administration setzte diese nicht konformen Biomaterialien bei Amerikanern ein, und Xavier Becerra unter Biden tut dies auch heute noch.
Der politische Druck.
Der folgende E-Mail-Austausch fand am 16. November 2020 zwischen leitenden Angestellten der EMA statt:
Noel Wathion – Stellvertretender Exekutivdirektor (geht im Juni 2021 in den Ruhestand):

Agnes Saint-Raymond – Leiterin der Abteilung für internationale Angelegenheiten:

Marco Cavaleri, Vorsitzender der Pandemie-Taskforce Covid-19 bei der EMA:

Die Emails sollten von unten nach oben gelesen werden.

Ein paar interessante Dinge: Die drei Aufsichtsbehörden – die US-amerikanische FDA, die britische MHRA und die EU-EMA – sind alle damit beschäftigt, den Zeitpunkt der Zulassung zu koordinieren, bevor eine formale Prüfung der Daten stattgefunden hat, bevor die beratenden Ausschüsse die Ergebnisse der klinischen Studien gesehen, diskutiert und darüber abgestimmt haben usw. Sie diskutieren über den Zeitplan, da die Daten KEINE Rolle dabei spielen, ob diese Produkte auf den Markt kommen oder nicht. Außerdem interagieren sie so, als ob es sich nicht um drei getrennte Behörden separater souveräner Nationen handelt, die gegenüber verschiedenen Steuerzahlern und der Aufsicht des Kongresses/Parlaments verantwortlich sind, sondern einfach um bürokratische Abteilungen, die bereits zu einer globalen Regierung verschmolzen sind. Schließlich wird sich die FDA „in die EUA stürzen“, da sie „von Azar gedrängt wird“ (Alex Azar – damaliger HHS-Minister) und „Trump die Fäden zieht“.
Viele Leute fragen mich, wie es möglich ist, dass Tausende von Menschen an dem Betrug beteiligt waren, der als „Covid-Pandemieabwehr“ inszeniert wurde – es ist doch nicht möglich, dass so viele Leute unter einer Decke stecken! Es war nicht notwendig, dass so viele eingeweiht waren. Hier ist sich Noel Wathion, ein leitender Mitarbeiter der EMA, entweder nicht bewusst, dass die Überprüfung der Daten für die Frage, ob die Injektionen letztendlich auf den Markt kommen, irrelevant ist, oder er stellt dies geschickt falsch dar (ich glaube sogar, dass er sich dessen nicht bewusst war). Die ihm unterstellten EMA-Mitarbeiter brauchen sich dessen nicht bewusst zu sein und müssen sich nur beeilen, um die ihnen zugewiesene Aufgabe zu erfüllen. Die Abschottung ist der Schlüssel zur Vertuschung eines großen Betrugs in großen Organisationen und komplexen Strukturen. Ist das der Grund, warum er kurz nach der Veröffentlichung der Killshots zurückgetreten/zurückgetreten ist? Er steht auch unter dem Druck der Europäischen Kommission, das Medikament zu genehmigen. Und Pfizer will jetzt eine vollständige Zulassung (MA) anstelle der bedingten Zulassung (CMA)! Anmerkung – die CMA wurde erteilt, aber die Bedingungen wurden von Pfizer/BioNTech nie erfüllt, denn wen interessiert das schon, es war von Anfang an ein Spiel.
„(Co)-Rapps“ = Mitberichterstatter. Die EMA ist eine europäische Einrichtung, die sich aus den ehemals getrennten „zuständigen Behörden“ der Mitgliedstaaten zusammensetzt, die früher die Arzneimittel in jedem Land separat regulierten und genehmigten. In der europäischen Struktur wird das technische Überprüfungs- und Co-Review-Team für ein bestimmtes Produkt ausgewählt. Im Fall der Covid-„Impfstoffe“ war das schwedische Team unter der Leitung von Philip Josephson der Berichterstatter (Hauptberichterstatter) und das französische Team unter der Leitung von Jean-Michel Race – der Mitberichterstatter. „CHMP“ =Ausschuss für Humanarzneimittel (bei der EMA).

Die E-Mail ist an Olga Solomon bei der Europäischen Kommission gerichtet, und Noels Vorgesetzte, Emer Cooke, geschäftsführende Direktorin der EMA und frühere leitende Angestellte der WHO, ist darin enthalten. Hier ist Emer Cooke:

Ursulas schlauer Pakt.
Erinnern Sie sich an sie?

Ursula von der Leyen – EU-Kommissarin, die unter anderem unglaubliche räuberische Pfizer-Lieferverträge im Namen aller EU-Mitgliedstaaten per Textnachricht mit Pfizer-Chef Albert Bourla ausgehandelt hat. In diesen Verträgen mussten die EU-Länder staatliche Vermögenswerte als Sicherheiten hinterlegen, auf alle Qualitätskontroll-, Import- und Verbraucherschutzgesetze verzichten und ihre nationale Souveränität aufgeben – d. h., sie durften die Gesetzgebung in Bezug auf die Impfstoffhaftung nicht durch ihre eigenen Parlamente ändern? Die räuberischen Verträge, die vollständig geschwärzt wurden, um die so genannten „kommerziellen Interessen von Pfizer“ zu schützen. Der folgende E-Mail-Austausch bezieht sich auf Ursulas tapfere Bemühungen:

Es werden eine Reihe von Akronymen verwendet, die wichtigsten sind „EC“ = Europäische Kommission, „MS“ = Mitgliedstaaten, „EP“ = Europäisches Parlament. Der Schlüsselsatz lautet, dass Ursula „bereit ist, die zuständigen Gesundheitsminister persönlich anzurufen, um die Anwendung von Artikel 5 Absatz 2 zu vermeiden“. Worum geht es hier? Artikel 5(2) bezieht sich auf „Artikel 5(2) der Richtlinie 2001/83“ – eine Notfallgenehmigung in einem europäischen Mitgliedstaat, die von jedem Mitgliedstaat separat in seinem eigenen Land erteilt wird. Die CMA ist eine bedingte Marktzulassung, die von der EMA für alle EU-Mitglieder gleichzeitig erteilt wird. Sie wird von der EU als ein weitaus robusteres Verfahren als eine EUA beworben (Hervorhebung von mir):
…Die CMA folgt einem kontrollierten und robusten Rahmen, der Garantien bietet, die eine Notfallzulassung nicht bieten kann. In Wirklichkeit handelt es sich bei einer Notverwendungsgenehmigung nicht um eine Zulassung des Impfstoffs, sondern um eine Genehmigung für die vorübergehende Verwendung des nicht zugelassenen Impfstoffs. Das CMA stellt sicher, dass alle Pharmakovigilanz- und Herstellungskontrollen, einschließlich Chargenkontrollen für Impfstoffe, sowie andere Verpflichtungen nach der Zulassung rechtsverbindlich gelten […]. Insbesondere:
-Es gewährleistet eine strenge Überwachung der Sicherheit des Arzneimittels in der gesamten EU durch das EU-Pharmakovigilanzsystem. […]
-Sie gewährleistet die Sicherheitsüberwachung nach der Zulassung und ermöglicht die strukturierte Erhebung zusätzlicher Daten. […].
-StrengeHerstellungsprozesse, einschließlich der Chargenfreigabe für Impfstoffe und den Vertrieb, unterliegen denselben laufenden Kontrollen wie alle zugelassenen Arzneimittel. Die Überwachung der Herstellungsprozesse stellt sicher, dass das Arzneimittel nach hohen pharmazeutischen Standards im Rahmen einer groß angelegten Vermarktung hergestellt und kontrolliert wird.
Im Rahmen einer bedingten EU-Zulassung für das Inverkehrbringen (CMA) liegt die Haftung beim Zulassungsinhaber. Der Zulassungsinhaber ist für das Produkt und seine sichere Anwendung verantwortlich.
In der Theorie klingt das großartig. Genau das hat Ursula versprochen, als sie persönlich bei den Politikern in den Mitgliedstaaten anrief und ihnen die Arme verdrehte. Vielleicht war das Armdrücken gar nicht nötig, da sie durch die Covid-Propaganda ausreichend verängstigt waren und auf die Wunder-„Impfstoffe“ warteten, um sie zu retten. Das Problem ist, dass Ursula nie vorhatte, diese Versprechen zu erfüllen, und dass es auf jeden Fall nicht möglich ist, die mRNA-„Impfstoffe“ in der für Arzneimittel erforderlichen Sicherheit, Wirksamkeit und Herstellungsqualität zu produzieren. Was Ursula von diesem Prozess wirklich brauchte, war, alle europäischen Mitgliedstaaten in einen Pakt einzubinden, indem sie eine „robuste“ CMA versprach, damit sie keine unabhängige Autorität über die in ihren Ländern vertriebenen Impfungen haben konnten. Der Weg über Artikel 5 hätte bedeutet, dass jeder Mitgliedstaat das Produkt genehmigen könnte und dann die Befugnis hätte, die Genehmigung zu widerrufen, wenn Probleme festgestellt würden. Artikel 5 sieht auch eine Haftungsfreistellung für den Hersteller vor, macht es aber unmöglich, das Produkt vorzuschreiben. Auf dem Weg über die CMA könnte keiner der Mitgliedstaaten eine unabhängige Entscheidungsbefugnis ausüben, und so könnte sie sie alle in die gleichen, unsinnigen und fast vollständig redigierten Verträge mit Pfizer, Moderna und AstraZeneca zwingen, die ohnehin auf jegliche Haftung verzichten und den Ländern darüber hinaus verbieten, ihre eigenen Gesetze in Bezug auf die Haftung zu ändern!
Die Käufer müssen Pfizer „entschädigen, verteidigen und schadlos halten … von und gegen alle Klagen, Ansprüche, Aktionen, Forderungen, Verluste, Schäden, Verbindlichkeiten, Vergleiche, Strafen, Bußgelder, Kosten und Ausgaben …, die sich aus dem Impfstoff ergeben, mit ihm in Verbindung stehen oder aus ihm resultieren.“
Bemerkenswerte Hauptbeanstandungen und deren Fehlen durch die EMA-Gutachter.
Der Abschnitt „Chemistry Manufacturing and Controls“ (CMC) des Zulassungsantrags für biologische Arzneimittel ist die wichtigste Säule der Zulassung. Er beschreibt den Herstellungsprozess und die Einhaltung der Guten Herstellungspraxis (cGMP) sowie eine Reihe von Gesetzen und Vorschriften, die die Reinheit, Wirksamkeit, Konsistenz und Sicherheit der serienmäßig hergestellten Arzneimittel und Biologika gewährleisten sollen. Die Sicherheits- und Wirksamkeitsdaten aus klinischen Studien sind nutzlos, wenn der Hersteller den Aufsichtsbehörden und der medizinischen Gemeinschaft nicht versichern kann, dass: 1) das fragliche Produkt gemäß der Spezifikation in klinischen Versuchen verwendet wurde, 2) das Produkt konsistent, rein, qualitativ hochwertig und reproduzierbar ist, mit gut charakterisierten und vorhersehbaren Herstellungsprozessen und Kontrollschritten, 3) das gleiche Produkt wie in den Versuchen kommerziell vertrieben wird.
Im CMC-Abschnitt des Pfizer-Antrags gab es Probleme:

Die CMC-Bewerter waren mit dem für die Bewertung angegebenen Zeitplan nicht zufrieden, da er alle normalen und auch alle beschleunigten Fristen bei weitem überschritt. Die Lösung bestand also darin, dass die Gutachter einfach „gedrängt“ werden mussten. Damit wurde nur ein Ziel erreicht: Menschen, die möglicherweise Bedenken hätten äußern können, an den Rand der Erschöpfung zu treiben, damit sie einfach aufgeben und mitmachen. Schließlich wussten die Verantwortlichen ganz genau, dass die behördliche Überprüfung keine Bedeutung und keine Auswirkung auf die gefälschte „Zulassung“ hatte, sie würde so oder so stattfinden. Im Vereinigten Königreich hat die MHRA bereits zugegeben, dass sie keine formale Befugnis zur Überprüfung und Genehmigung dieser Injektionen hatte, und ich bin bereit zu wetten, dass auch die EMA keine solche Befugnis hatte.
Die CMC-Bewerter auf unterer Ebene wussten dies nicht und arbeiteten sehr hart und höchstwahrscheinlich in gutem Glauben. Bis Ende November erhoben sie mehr als 140 formale Einwände gegen den CMC-Antrag von Pfizer, der immer noch viele Lücken und fehlende Informationen aufwies. Bei 10-15 behördlichen Einwänden kann ein pharmazeutischer Antrag normalerweise nicht weiter bearbeitet werden, bis die Einwände ausgeräumt sind. Drei wichtige Einwände, d. h. formale rote Fahnen, werden in den nachstehenden E-Mails ausdrücklich erwähnt. Ich und andere haben ausführlich über MO#2 (fehlende mRNA-Integrität) geschrieben. Hier finden Sie eine E-Mail von Evdokia Korakianiti, einer der Gutachterinnen, und eine Antwort von Alexis Nolte, in der das Problem und die (völlig unbekannten und möglicherweise sehr beunruhigenden) Auswirkungen auf die Wirksamkeit und Sicherheit des Produkts erörtert werden:

Das Problem des mRNA-Abbaus wurde auch von den CMC-Qualitätsspezialisten Ton van der Stappen, Senior Biopharmaceutical Expert beim Medicines Evaluation Board (mit Sitz in den Niederlanden) und Qualitätsspezialist für die EMA, diskutiert:

und Brian Dooley, ein weiterer pharmazeutischer Qualitätsspezialist bei der EMA:

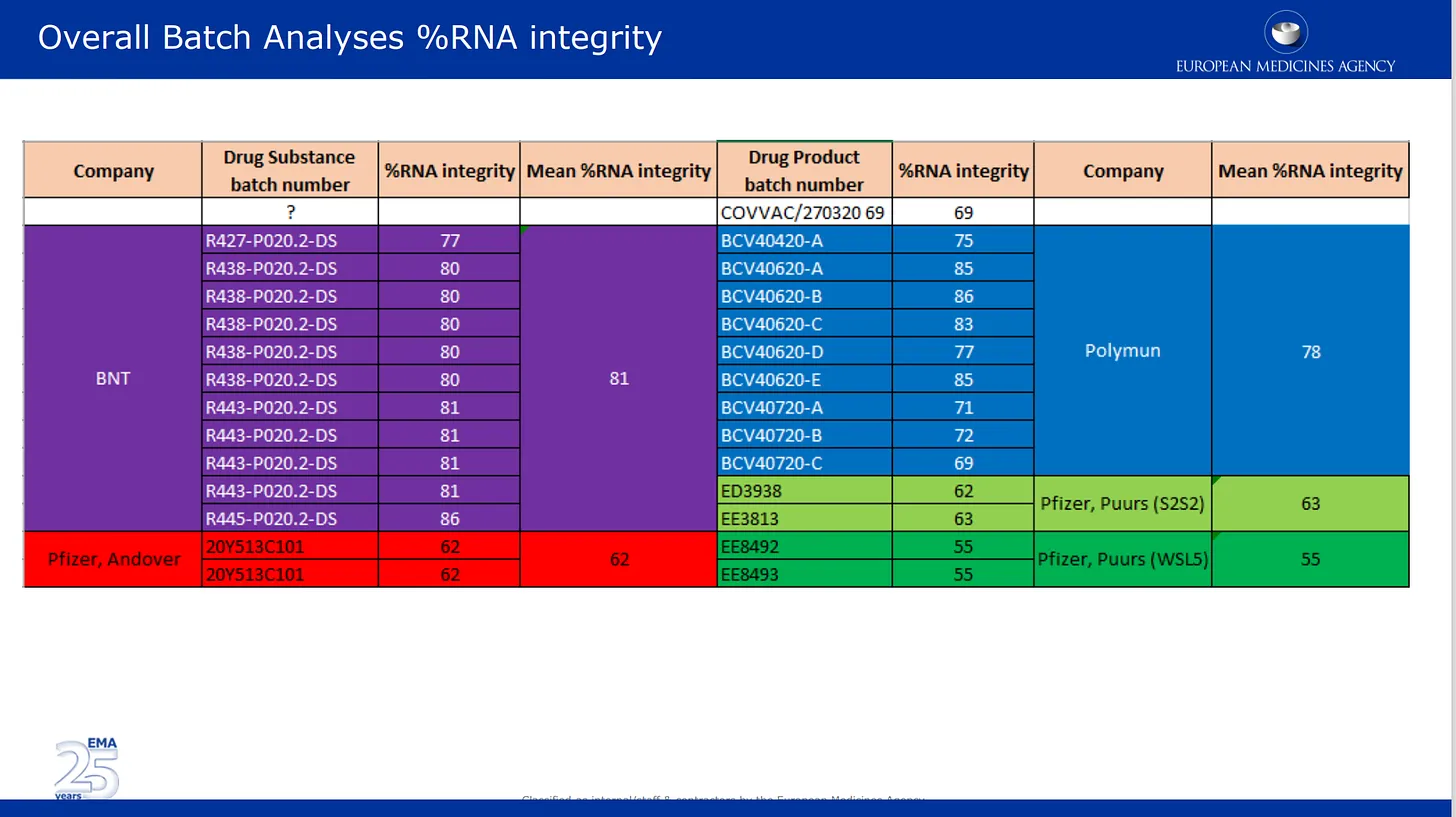
Die folgenden Bilder stammen aus der Qualitätsprüfung, die sie der EMA vorgelegt haben. Im ersten Bild geht es um das inzwischen gut dokumentierte Problem des mRNA-Abbaus in verschiedenen Chargen des Produkts von Pfizer. Hier sind die von Pfizer gelieferten Ergebnisse der Chargenanalyse nach den verschiedenen Herstellungsstandorten sowie nach Wirkstoff und Arzneimittelkategorie aufgelistet und farblich kodiert. Arzneimittelsubstanz = aktive Komponenten des Produkts (mRNA allein) und Arzneimittelprodukt ist die in den Lipiden und anderen Bestandteilen formulierte Substanz. Der Prozentsatz der mRNA-Integrität beschreibt den Prozentsatz der in einer Charge nachgewiesenen mRNA in voller Länge. Der andere Teil der Charge bestand aus unbekannten Bruchstücken mit unbekannten Eigenschaften oder Auswirkungen auf die Sicherheit. Es sei darauf hingewiesen, dass die Aufsichtsbehörden keine unabhängige Prüfung dieser Angaben vorgenommen haben, sondern lediglich die von Pfizer/BioNTech angegebenen Zahlen aufführten.
Es scheint, dass diese beiden wissenschaftlichen Berater die gefälschten Bilder der Western-Blot-Ergebnisse, die Pfizer der EMA vorgelegt hat,geprüft und akzeptiert haben – hier sind sie in ihrer eigenen PowerPoint-Präsentation vom 24. November 2020 zu sehen. Lesen Sie die Notiz unter der Folie – sie akzeptieren diese Bilder als echt, obwohl die beiden Gutachter es besser wissen müssten. Warum haben sie das NICHT beanstandet? So steht es in der Notiz:
Die Größe des Proteins nach In-vitro-Expression des Wirkstoffs BNT162b2 wurde mittels Western Blot bestimmt. Es wurde bestätigt, dass die Größe des exprimierten Proteins bei den drei Prozess-1-Chargen und der Prozess-2-Charge vergleichbar ist. Abbildung 3.2.S.2.6-15 zeigt, dass die Größe des exprimierten Proteins mit der erwarteten Größe des Wirkstoffs BNT162b2 übereinstimmt und mit allen getesteten Chargen vergleichbar ist. Darüber hinaus sind die relativen Expressionsniveaus für alle Chargen vergleichbar, was durch eine vergleichbare Bandenintensität bei jedem Belastungsniveau in allen Chargen belegt wird.

Vielleicht sollten sich einige hartgesottene Journalisten an die Herren Dr. van der Stappen und Dooley sowie an Frau Korakianiti und andere hier erwähnte Personen wenden und um eine Stellungnahme bitten.
Aus den Antworten von Evdokia Korakianiti geht hervor, dass die EMA-Leitung auf Waffen verzichtete und sich auf „Daten stützte, die nur die FDA gesehen hat“, aber „optimistisch“ ist und dass die FDA behauptete, mRNA-Bruch sei ein „theoretisches Problem“. Wirklich? Gibt es Daten, die diese Aussage stützen oder nicht? Hier sind die E-Mails, aus denen hervorgeht, dass die wesentlichen Einwände formell aufgeschrieben und anschließend von der EMA ignoriert wurden, da das Produkt nur ein paar Wochen später kommerziell ausgeliefert wurde. Die Bedingungen der CMA wurden nie erfüllt.
Dies bestätigt, was wir bereits wissen: Weder die EMA (noch die FDA, Health Canada, MHRA oder andere Aufsichtsbehörden) hatten wirkliche Befugnisse über diese Produkte oder Einfluss darauf, ob sie in der ahnungslosen Öffentlichkeit eingesetzt werden. Es war von Anfang bis Ende alles nur Theater.
Hier sind die drei wichtigsten Einwände, die bis heute nicht ausgeräumt sind:

Und hier wird mit den Händen gewedelt und die Behauptungen der FDA ohne Hinterfragung oder formale Bewertung der Daten durch die EMA-Behörden übernommen:

Abschließend kann ich sagen, dass ich ungefähr 70 verschiedene Personen gezählt habe, die in dem durchgesickerten Dokument und in den E-Mails erwähnt werden und die diese tragische Scharade ermöglicht haben – die „Zulassung“ des tödlichsten Produkts, das jemals auf die größte Anzahl von Menschen losgelassen wurde, was zu einer noch nie dagewesenen Zahl von Toten und Verletzten weltweit führte. Vielleicht wurden die meisten von ihnen, von einigen Ausnahmen abgesehen, im Jahr 2020 getäuscht und haben nicht verstanden, dass sie an einem Kriegsverbrechen beteiligt waren und einen tödlichen Betrug mit ihrer Unterschrift bestätigten. Ich glaube, dass die meisten von ihnen inzwischen Bescheid wissen, und ich hoffe, dass sie hinreichend entsetzt sind über das, was sie ermöglicht haben, und ich hoffe, dass diese Menschen als Whistleblower hervortreten und anfangen zu reden. Wir brauchen Antworten.
Ursprünglich veröffentlicht von Due Diligence and Art
Korrektur vorschlagen




