

Vaccins Covid-19 : les e-mails et documents de l’EMA révèlent des préoccupations majeures concernant l’intégrité des lots Pfizer et la course à l’autorisation de mise sur le marché
Notre récent article de Meryl Nass, MD, sur le déploiement de vaccins le plus rapide de l’histoire, détaillant la procédure d’approbation des nouveaux » vaccins » à ARNm, met en évidence l’incompréhensible ignorance du fonctionnement des autorités sanitaires aujourd’hui.
Dans le même temps, la commission spéciale du Parlement européen sur la pandémie de COVID-19 a eu l’occasion de poser des questions aux représentants de quatre sociétés pharmaceutiques le 5 septembre 2022. La réunion a porté sur les activités passées et futures liées à la pandémie et quelques députés européens ont tenté d’apporter de la transparence et la responsabilité attendues dans un cadre démocratique. La vidéo ci-dessous présente les questions posées par M. Cristian Terhes, membre du Parlement européen.
Pour tous ceux qui pourraient être choqués par le déploiement rapide des nouveaux « vaccins » en Europe, nous devons nous rappeler et nous interroger sur la manière dont s’est déroulée l’autorisation de la campagne de vaccination contre le COVID-19, comme nous l’avons appris par des fuites d’e-mails de l’EMA (Agence européenne des médicaments), résumées dans l’article suivant de la journaliste d’investigation Sonia Elijah, sur Trial Site News (juin 2022).
Vaccins Covid-19 : les e-mails et documents de l’EMA révèlent des préoccupations majeures concernant l’intégrité des lots Pfizer et la course à l’autorisation de mise sur le marché
par Sonia Elijah
Trial Site News (vidéo ci-dessus) a récemment pu consulter des e-mails internes de l’Agence européenne des médicaments (EMA) et un compte rendu de réunion entre l’agence et Pfizer. L’EMA supervise l’évaluation et la surveillance des médicaments pour l’Union européenne. Comme d’autres organismes de réglementation sanitaire, sa principale responsabilité est de protéger et de promouvoir la santé publique. Des extraits de la correspondance électronique interne de l’EMA, une présentation PowerPoint du 26 novembre 2020 d’une réunion cruciale entre Pfizer et l’agence, ainsi qu’un rapport confidentiel de 43 pages de Pfizer ont été fournis par une source anonyme en raison de sa confiance dans l’engagement de Trial Site News en faveur de la transparence, accessibilité de l’information et responsabilité afin de garantir l’éthique et la qualité d’une industrie de la recherche biomédicale centrée sur la santé publique.
Les organismes de réglementation, comme l’EMA, la Food and Drug Administration (FDA) aux États-Unis et la Medicines and Healthcare products Regulatory Agency (MHRA) au Royaume-Uni, ont pour mission de prendre des décisions dans l’intérêt du public. Les influences extérieures, telles que la pression politique ou médiatique, ne sont pas censées être un facteur déterminant dans leur prise de décision. Toutefois, lorsqu’il s’est agi d’une pandémie et de l’autorisation de mise sur le marché conditionnelle accélérée des vaccins Covid-19 (en particulier pour les vaccins à base d’ARNm produits par Pfizer-BioNTech et Moderna), il semble que ces derniers l’aient emporté.
La correspondance électronique dont il est question date du 10 au 25 novembre 2020, quelques semaines seulement avant que l’EMA n’accorde l’AMC (autorisation de mise sur le marché conditionnelle) pour le vaccin Covid-19 de Pfizer-BioNTech le 21 décembre 2020. La FDA a accordé l’EUA (autorisation d’utilisation d’urgence) pour ce vaccin le 11 décembre et la MHRA a été la première à franchir la ligne d’arrivée le 2 décembre. L’auteur utilise ici le terme » ligne d’arrivée « , car les e-mails révèlent une course intense, presque compétitive, pour autoriser les vaccins Covid-19 le plus rapidement possible. Il est évident qu’à l’époque, le monde était en proie à une pandémie et qu’il y avait un grand besoin d’autoriser un vaccin pour protéger les gens contre le nouveau coronavirus.
La ruée vers EUA (autorisation d’utilisation d’urgence)
Dans un courriel de Marco Cavaleri, à l’époque responsable des menaces biologiques pour la santé et de la stratégie en matière de vaccins à l’EMA, il communique avec urgence comment la FDA américaine « va se précipiter sur l’EUA ».
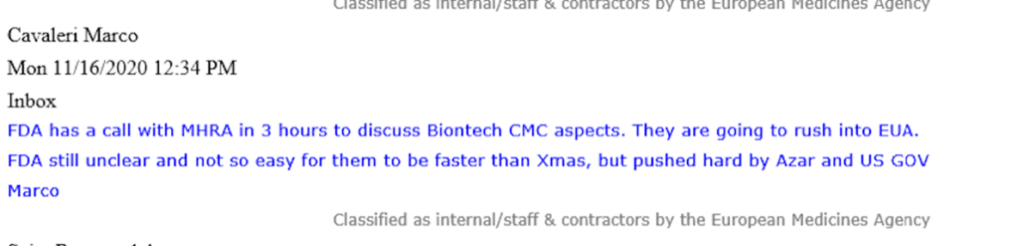
Cavaleri fait référence à cette « ruée » (warp speed) qui est « poussée durement par Azar et US GOV« . Sous l’administration Trump, Alex Azar, ancien cadre de l’industrie pharmaceutique, a été le secrétaire américain à la santé et aux services sociaux (HHS) de 2018 à 2021. La FDA est une agence qui relève directement du HHS.
Il convient de noter que lorsque Azar était l’ancien président de Lilly USA LLC, une division d’Eli Lilly, les prix des médicaments ont explosé sous sa direction. La société pharmaceutique a également été impliquée dans une action collective sous son mandat, accusée d’avoir exploité le système de fixation des prix des médicaments pour augmenter les bénéfices de son médicament contre l’insuline. Bien sûr, cela ne signifie pas nécessairement que ce dirigeant était complice de quelque manière que ce soit, mais le moment choisi est remarquable.
L’e-mail de Cavaleri montre à quel point la politique (et le gouvernement américain) dirigeait le processus de réglementation de la FDA, en s’assurant qu’il se déroulait à la » vitesse de la lumière « . Et bien sûr, sur cette note, l’opération Warp Speed de Trump devait garantir que tous les records de développement de vaccins seraient pulvérisés. Les intentions étaient sans doute bonnes compte tenu de l’apparition de la pire pandémie depuis un siècle.
Cependant, de l’autre côté de l’Atlantique, au sein de l’agence de réglementation européenne, la tension est montée d’un cran alors que la pression exercée pour accélérer les délais rendait l’air et l’humeur générale tendus – la pression et l’anxiété étaient palpables dans les échanges de courriels examinés.
Des personnes d’une grande intégrité et d’une grande clarté quant à leurs rôles et engagements en tant que gardiens de la santé publique sont apparues. Par exemple, une personne s’est montrée très préoccupée par l’accélération des délais pour s’assurer de respecter la » date limite » d’autorisation des vaccins, au détriment d’une évaluation solide. Il s’agissait de Noel Wathion, à l’époque directeur exécutif adjoint de l’EMA, mais qui a depuis pris sa retraite. Ce fonctionnaire de l’EMA a souligné que « nous accélérons autant que possible, mais nous devons également nous assurer que notre évaluation scientifique est aussi solide que possible. N’oublions pas la responsabilité et l’obligation de rendre des comptes liées à la recommandation faite à la CE d’accorder une AMC.

Wathion suppose que l’EUA de la FDA (et de la MHRA) serait délivrée avant que l’EMA n’accorde sa propre AMC, ce qui s’est avéré exact. Ce qui est intéressant, c’est qu’il s’est soucié de limiter les dommages résultant de la probabilité que l’EMA finisse dernière dans cette course réglementaire et qu’il craint que l’opinion publique et les médias se retournent contre l’agence. La rapidité semble avoir supplanté les préoccupations de qualité si l’on en croit un examen attentif de ces e-mails.
Dans un courriel du 19 novembre, Wathion révèle une téléconférence « plutôt tendue » avec la commissaire européenne (Ursula von der Leyen) qui était « parfois même un peu désagréable« . Cela reflète la pression croissante à laquelle le personnel de l’EMA était soumis pour délivrer rapidement des AMC à la suite d’une autorisation européenne accordée par la FDA/MHRA pour le vaccin Pfizer-BioNTech. Von der Leyen est impliquée comme étant potentiellement responsable de cet environnement tendu avec » un retard de plusieurs semaines… difficilement acceptable pour la CE [Commission européenne] « .
Au début de l’année 2022, Trial Sites News a rapporté comment Mme von der Leyen s’est retrouvée au cœur d’un scandale lorsqu’un groupe de députés européens indépendants a exigé sa démission immédiate et la divulgation complète d’une série de SMS privés entre elle et le PDG de Pfizer, Albert Bourla. Seule une petite partie de ces textes a été divulguée. Parmi ceux qui l’ont été, ils révèlent qu’elle a négocié des parties d’un accord européen sur les vaccins, de manière unilatérale avec Bourla par le biais d’une série de textos ! Il est clair que les protocoles standard en Europe ont été jetés par la fenêtre en faveur de la rapidité et que cela semble être lié à une pression concurrentielle unifiée sur les trois agences de réglementation.
Wathion expose ses réflexions après ce CT particulier, et écrit de manière choquante que « les retombées politiques semblent trop importantes, même si le niveau « technique » des EM [États membres] pourrait défendre un tel délai afin de rendre le résultat de l’examen scientifique aussi solide que possible ». En d’autres termes, la science est d’abord apparue comme une couverture pour la politique suivie.

Wathion souligne qu’un retard potentiel de plusieurs semaines pour garantir « une assurance solide, en particulier en ce qui concerne la CMC et la sécurité » sera accueilli par « des critiques de diverses parties », notamment les médias, la CE (Commission européenne) et le PE (Parlement européen). Wathion dit craindre que si la date limite « pour s’aligner autant que possible sur le calendrier d' »approbation » de la FDA/MHRA » ne peut être respectée, « nous serons submergés sur tous les fronts et au milieu de la tempête« . Toutefois, ce retard potentiel semble être nécessaire « pour que le résultat de l’examen scientifique soit aussi solide que possible« . Cela implique que la rapidité au détriment de la sécurité était à l’ordre du jour pour éviter les « retombées politiques« . Il est clair le protocole d’autorisation du vaccin Covid-19 était dicté par la politique, et non la science.

Dans l’e-mail de Marco ci-dessus, le fonctionnaire de l’EMA révèle que le PDG de Pfizer, Albert Bourla, a fait du lobbying auprès de Peter Marks, ce qui pourrait être interprété comme très controversé, étant donné que Marks est le directeur du Center for Biologics Evaluation and Research (CBER) de la FDA. L’accès apparent de Pfizer au chien de garde fédéral soulève pour le moins des questions importantes, si ce n’est qu’il introduit la possibilité d’imbroglios inquiétants entre l’industrie et une agence fédérale scientifique prétendument indépendante.
Des préoccupations majeures quant à l’intégrité entre les lots de vaccins
Un e-mail de Cavaleri (voir ci-dessous) révèle qu’à l’époque, la FDA était au courant de « certains problèmes » liés aux CMC qui devaient être résolus et qui pourraient « finir par être la partie difficile« . Le terme CMC désigne la chimie, la fabrication et les contrôles, également appelés qualité pharmaceutique, qui couvrent diverses procédures utilisées pour évaluer et garantir la sécurité et la cohérence entre les lots de produits pharmaceutiques.
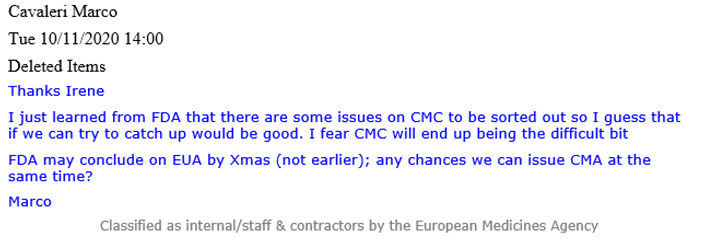
Un courriel d’Evdokia Korakianiti (administratrice scientifique de l’EMA) explique plus en détail ce que sont ces « problèmes » et comment il s’agit en fait de préoccupations majeures concernant le vaccin Pfizer-BioNTech.

De manière alarmante, des différences significatives dans les niveaux d’intégrité de l’ARNm entre les lots de vaccins commerciaux (à grande échelle) et cliniques (à petite échelle) de Pfizer-BioNTech ont été observées. L’intégrité de l’ARNm était de « ~78% » dans les lots cliniques et de « ~55% dans les lots commerciaux proposés « , la » cause fondamentale » n’ayant pas encore été identifiée. Les implications en matière de sécurité et d’efficacité dues à ce problème ont également été notées dans l’e-mail « comme devant être définies ».
Dans un rapport confidentiel de Pfizer, qui a également fait l’objet d’une fuite avec les courriels de l’EMA, la société déclare que selon l’expérience générale d’Acuitas Therapeutics (la société de biotechnologie qui a développé la plateforme de nanoparticules lipidiques pour le vaccin de Pfizer et Moderna), » un seuil minimum est d’environ 70 % « . (Voir la capture d’écran ci-dessous).

Ensuite, à la page 30, il est dit : « L’efficacité du produit dépend de l’expression de l’ARN délivré, qui nécessite une molécule d’ARN suffisamment intacte« . (Voir la capture d’écran ci-dessous)

Cette phrase exacte « nécessite une molécule d’ARN suffisamment intacte » a été utilisée dans l’e-mail d’Evdokia Korakianiti, membre du personnel de l’EMA, que j’ai inclus ci-dessus, envoyé le 23 novembre 2020 – maintenant nous savons probablement d’où Korakianiti l’a référencé.
Le fait que les lots commerciaux (qui allaient être distribués dans le monde entier) présentent un niveau d’intégrité de l’ARNm (molécule d’ARN intacte) aussi faible est très préoccupant étant donné son lien intrinsèque avec l’efficacité et la sécurité potentielle du produit.
Le lendemain, Veronika Jekerle, responsable du bureau de la qualité de la pharmacie, écrit à Evdokia (voir ci-dessous).

La différence dans le niveau d’intégrité de l’ARNm a de nouveau été notée comme une préoccupation majeure » partagée par la plupart des États membres » et son » impact potentiel sur la sécurité « . Jekerle souligne en gras, « Une approbation d’ici la fin de l’année pourrait potentiellement être possible, si ces préoccupations + GMP seront résolues. »
D’où la question cruciale : comment toutes ces préoccupations ont-elles pu être résolues alors que l’AMC a été accordée seulement quelques semaines plus tard, le 21 décembre ? Une manière possible de résoudre ce problème est expliquée plus loin dans ce rapport.
Contrairement aux préoccupations de certains autres responsables de l’EMA, Marco Cavaleri écrit à peu près au même moment dans l’mail suivant (voir ci-dessous) que le contenu en ARNm n’est pas une préoccupation majeure, selon la FDA – « la question du contenu en ARNm n’est pas perçue comme majeure« . Il déclare également, de manière choquante, qu’il n’est pas clair si des inspections BPC ont été effectuées. Cette révélation est très préoccupante étant donné que les BPC font référence aux bonnes pratiques cliniques, qui sont « une norme de qualité éthique et scientifique internationale pour la conception, la conduite, l’enregistrement et le compte rendu des essais impliquant la participation de sujets humains« .
Ce qui est encore plus alarmant, c’est la déclaration suivante : « aucun intérêt majeur de la part de la FDA « , qui semble révéler le manque apparent de préoccupation ou même d’intérêt de l’agence de réglementation quant à la réalisation des inspections des BPC dans le contexte des essais cliniques de Pfizer, sur lesquels la FDA s’est appuyée pour délivrer l’EUA pour le vaccin Pfizer-BioNTech. Dans l’un des précédents rapports d’enquête de cet auteur pour Trial Site News, nous avons noté que la FDA n’avait inspecté que 1% des sites d’essais de Pfizer.

D’autres informations accablantes sont révélées (voir la capture d’écran ci-dessous) lorsque plusieurs organismes de réglementation : Santé Canada (SC), l’EMA, la MHRA et la FDA sont tous conscients du problème de l’intégrité du % d’ARNm, mais la FDA et Santé Canada affirment sans preuve que « les problèmes de sécurité associés… sont plus théoriques« .

Santé Canada semble ensuite se contredire car il est décrit plus tard comme étant particulièrement préoccupé par le fait qu’une région reçoive « tout le matériel sous-optimal ». De toute évidence, il ne voulait pas être cette région.
Il est choquant de constater que la fin de l’e-mail révèle que le « demandeur [Pfizer] a partagé avec la FDA et nous [EMA]/MHRA seulement aujourd’hui et le problème des particules visibles dans le DP [produit pharmaceutique] semble être des composants de nanoparticules lipidiques)« .
Ceci est très inquiétant car ce problème important a été porté à la connaissance des trois principales agences de réglementation le 25 novembre, quelques semaines seulement avant que l’EMA n’accorde l’AMM et la FDA l’EUA pour le vaccin de Pfizer. De façon alarmante, c’était quelques jours seulement avant que la MHRA n’accorde l’autorisation au Royaume-Uni, le 2 décembre 2020. L’hypothèse de Veronika selon laquelle les « particules visibles » pourraient être des LNP (nanoparticules lipidiques) est difficile à accepter étant donné que les nanoparticules ne sont pas visibles à l’œil nu. D’autres anomalies étaient apparentes, mais il s’agissait probablement d’un effort historique en termes de rapidité de développement du vaccin. Il semble toutefois évident qu’un peu plus de temps était nécessaire.
Comment le % d’intégrité de l’ARNm a été apparemment résolu
L’écart entre les lots semble avoir été résolu lorsqu’il est mentionné que les » derniers lots [reçus par la FDA] indiquent que le % d’ARN intact est revenu autour de 70-75 % « .
Cependant, dans un rapport divulgué d’une réunion avec Pfizer et l’EMA le 26 novembre 2020, un jour après l’email de Veronika, il est révélé de manière choquante que la spécification de l’intégrité de l’ARN a été révisée à la baisse à >=50% pour la durée de conservation du médicament, ce qui est nettement inférieur au seuil minimum de 70% qu’Acuitas Therapeutics avait stipulé et à la moyenne de 78% des lots cliniques. Est-ce la façon dont l’EMA (et potentiellement la FDA/MHRA/HC) a » résolu » le problème pour garantir « une approbation avant la fin de l’année » ?

Il est fait mention « d’incertitudes quant à la constance de la qualité du produit et donc d’incertitudes quant à la sécurité et à l’efficacité du produit commercial« . Pourtant, il est difficile de comprendre comment l’abaissement de la spécification d’intégrité de l’ARN pourrait remédier à cette objection majeure.
Dans une autre diapositive, l’artefact indique que « les espèces d’ARN tronquées [raccourcies] et modifiées doivent être considérées comme des impuretés liées au produit« . Cela confirme que ces espèces d’ARNm raccourcies qui ont réduit le niveau d’intégrité du % d’ARNm ont été classées comme des impuretés. Une autre préoccupation alarmante découlant de ces impuretés est signalée par « la possibilité de protéines traduites autres que la protéine de pointe prévue (S1 S2) résultant d’espèces d’ARNm tronquées et/ou modifiées doit être examinée« . (Voir la capture d’écran ci-dessous)

Les preuves contenues dans ce rapport confirment que les organismes de réglementation tels que la FDA, la MHRA, l’EMA et Santé Canada étaient au courant des différences entre les lots en ce qui concerne le pourcentage d’intégrité de l’ARNm et que, de ce fait, l’effet sur la « sécurité et l’efficacité » était inconnu. Le rapport de réunion de Pfizer/EMA qui a fait l’objet d’une fuite soulève des préoccupations importantes en supposant que le problème a été résolu en abaissant simplement la spécification de l’intégrité de l’ARNm. En d’autres termes, le problème n’a peut-être jamais été résolu.
Le site web howbadismybatch.com, qui traite de la différence entre les lots de vaccin Covid, a attiré beaucoup d’attention. Il s’agit d’une base de données complète contenant des analyses sur les « codes de lot et les décès, handicaps et maladies associés aux vaccins Covid 19« . En saisissant le numéro de lot de n’importe lequel des vaccins Covid-19, on obtient la fréquence des événements indésirables signalés associés à ce lot.
J’ai parlé avec Sasha Latypova, qui a mené des essais cliniques pendant plus de 25 ans et possède sa propre société de biotechnologie, pour lui demander son avis d’expert sur les documents qui ont fait l’objet d’une fuite. Elle a répondu :
« Le manque d’intégrité de l’ARNm et la présence de fragments d’ARN non caractérisés dans les lots du produit de Pfizer ont été identifiés comme une « objection majeure » – un drapeau rouge réglementaire officiel, considéré comme une impureté du produit et qui aurait été un obstacle dans tout processus normal d’approbation d’un médicament. Il aurait fallu au moins un essai clinique supplémentaire pour évaluer les effets cliniques, ce qui aurait pris des mois à concevoir et à mener correctement. La panique a pris le pas sur l’intégrité scientifique et une norme d’acceptation des lots arbitrairement abaissée a été adoptée dans le but de respecter un délai motivé par des considérations politiques. À ce jour, cette question n’est toujours pas résolue et pourrait être la cause sous-jacente de l’énorme variation des taux d’événements indésirables et de décès observés pour différents numéros de lots de fabrication dans le VAERS du CDC et d’autres bases de données.«
Mme Latypova a fait une référence pertinente au sort du Titanic, en établissant une comparaison avec la façon dont les organismes de réglementation ont mené leur processus d’autorisation des vaccins Covid-19 à la vitesse de la lumière. Le capitaine du Titanic, Edward J. Smith, cherchait à améliorer le temps de traversée d’un navire, ce qui signifiait que le navire voyageait beaucoup trop vite, dans des eaux connues pour être couvertes de glace. Cela a provoqué une collision fatale avec un iceberg et le reste appartient à l’histoire.
À la lumière des preuves incluses dans ce rapport et du fait que le vaccin Covid-19 de Pfizer-BioNTech est l’un des produits les plus lucratifs de l’histoire (l’année dernière, Pfizer a réalisé un chiffre d’affaires de 37 milliards de dollars, les prévisions pour 2022 étant de 32 milliards de dollars), l’auteur s’efforce d’ouvrir le débat en posant certaines questions essentielles auxquelles doivent répondre les organismes de réglementation concernés, Pfizer et les membres de la communauté scientifique/médicale :
Quelles sont les implications en termes de sécurité et d’efficacité d’une intégrité de l’ARNm considérablement réduite (résultant d’un ARNm tronqué et modifié) dans les lots commerciaux de ce vaccin ?
Quelles sont exactement les particules visibles observées dans le DP (produit pharmaceutique) que Pfizer a partagé à la dernière minute avec l’EMA, la FDA et la MHRA et quelles en sont les implications en termes de sécurité et d’efficacité ?
Les réponses à ces questions sont d’une importance publique majeure.
Publié originellement sur Trial Site News.
À voir également : Interview de Sonia Elijah par Reiner Fuellmich au comité d’enquête Corona ici.






