Cartas desde el Inframundo
Crédito de la imagen: Shutter Stock
Los correos electrónicos filtrados de la EMA
Por Sasha Latypova
A finales de 2020 se filtró una colección de documentos -aproximadamente 900 páginas- de la sección de Fabricación Química y Controles (CMC) de la «vacuna» de Pfizer de la presentación reglamentaria a la Agencia Europea de Medicamentos (EMA), que se enviaron a varios periodistas. Los documentos también incluían intercambios de correos electrónicos entre algunos de los revisores y altos ejecutivos de la EMA. El British Medical Journal se hizo eco de la filtración y confirmó que los documentos eran auténticos. Recibí estos documentos de un colega aproximadamente un año después, a finales de 2021, y leí y utilicé muchos de ellos en mis análisis. He visto muchos documentos relacionados con I+D de Pfizer en mi trabajo profesional, por lo que también puedo confirmar que estos filtrados eran muy coherentes con la documentación típica de Pfizer. La EMA no negó la autenticidad, y sólo afirmó que se habían cambiado los encabezados de algunos de los correos electrónicos.
Ahora tengo un número mucho mayor de seguidores y una comprensión mucho más clara de la organización y la estructura pseudo-legal del cártel criminal que dirige la atrocidad global coloquialmente conocida como la «respuesta a la pandemia covid 19». Estoy revisando los correos electrónicos filtrados, ya que creo que proporcionan algunas pruebas muy significativas.
Los archivos de correo electrónico de la EMA que he leído contienen 14 capturas de pantalla de correos electrónicos de mediados a finales de noviembre de 2020. Los intercambios son del personal de la EMA y de altos ejecutivos. En mi opinión estos correos electrónicos demuestran que:
- Los revisores de la EMA estaban bajo una presión política masiva para inventar nuevas formas de aprobar estos productos peligrosos inaprobables. La presión emanaba de las altas esferas de los gobiernos de EE.UU., Reino Unido y la UE.
- La Comisaria de la UE, Ursula vonden Leyen, hizo promesas a los Estados miembros que nunca tuvo intención de cumplir con el fin de vincularlos a todos en un pacto único en relación a los contratos de vacunas y adelantarse así a cualquier decisión independiente en sus propios países.
- Hubo problemas graves e irresolubles -dado el calendario intencionadamente poco realista- con la calidad del producto que el personal de la EMA se vio presionado a aprobar. Algunos se sentían incómodos haciéndolo y expresando sus preocupaciones. Otros «pasaron por alto» datos claramente inventados.
En última instancia, la propia revisión reglamentaria y las preocupaciones planteadas no importaron: el producto iba a comercializarse a pesar de todo. Ahora sabemos exactamente por qué: las autoridades reguladoras no tenían poder regulador sobre él. Los reguladores farmacéuticos no supervisan los materiales militares conocidos como «contramedidas» y «demostraciones de fabricación» (lenguaje evasivo que encubre los agentes de guerra biológica fabricados por el Gobierno estadounidense capturado y sus socios mundiales). Los correos electrónicos muestran que la mayoría del personal de la EMA eran actores involuntarios en esta obra.
La confirmación de esto por el Reino Unido salió a la luz recientemente.
Basado en la respuesta a MHRA FOIA:
«Todas las vacunas Covid y las decisiones de autorización terapéutica fueron tomadas por el Ministro de Licencias y no fueron delegadas»
Traducción: normalmente, el Secretario de Estado de Sanidad (Reino Unido) delega formalmente en la MHRA la autoridad para revisar y aprobar nuevos productos farmacéuticos. En el caso de los productos con relación al covid, la delegación de autoridad no existe. Parece que todos ellos fueron desplegados en solitario por Matt Hancock (aunque él señala con el dedo a alguien de más arriba). Lo mismo sucedió en los EE.UU. – Alex Azar bajo la Administración Trump desplegó estos materiales biológicos no conformes con regulaciones en los estadounidenses, y Xavier Becerra bajo Biden sigue haciéndolo hoy.
La presión política.
El siguiente intercambio de correos electrónicos se produjo el 16 de noviembre de 2020 entre altos ejecutivos de la EMA:
Noel Wathion – Director Ejecutivo Adjunto (jubilado en junio de 2021):

Agnes Saint-Raymond – Jefa de la División de Asuntos Internacionales:

Marco Cavaleri, Presidente del Grupo de Trabajo sobre Pandemia Covid-19 de la EMA:

Los correos deben leerse de abajo arriba.

Un par de cosas interesantes: los tres organismos reguladores -la FDA de EE.UU., la MHRA del Reino Unido y la EMA de la UE- están ocupados coordinando el calendario de aprobación antes de que se haya producido cualquier revisión formal de los datos, antes de que los comités consultivos hayan visto los resultados de los ensayos clínicos, los hayan debatido, votado sobre ellos, etc. Están discutiendo el calendario, ya que los datos NO IMPORTAN si estos productos van a salir al mercado o no. Además, están interactuando como si no fueran tres agencias separadas de naciones soberanas separadas responsables ante conjuntos separados de contribuyentes y supervisión del Congreso/parlamento, sino simplemente departamentos burocráticos ya fusionados en un gobierno global. Por último, la FDA va a «precipitarse en la EUA», siendo «empujada por Azar» (Alex Azar – Secretario del HHS en ese momento) y «Trump está moviendo los hilos».
Mucha gente me pregunta cómo es posible que miles de personas participaran en la estafa orquestada como «respuesta a la pandemia del covid» – ¡seguramente no es posible tener a tanta gente confabulada! No era necesario tener a tantos al tanto. En este caso, Noel Wathion, un alto ejecutivo de la EMA, o bien no es consciente de que la revisión de los datos es relevante para la comercialización de las inyecciones, o bien lo tergiversa hábilmente (de hecho, creo que no era consciente). Por lo tanto, el personal de la EMA que está por debajo de él no tendría por qué ser consciente, y simplemente se apresuraría a hacer cualquier estrecha tarea que se les asignará. La compartimentación es clave para encubrir cualquier gran estafa dentro de grandes organizaciones y estructuras complejas. ¿Por eso dimitió/se jubiló poco después del comienzo de la introducción de estas vacunas tran problemáticas? También está bajo presión de la CE (Comisión Europea) para aprobarlo. ¡Y Pfizer ahora quiere una Autorización de Comercialización completa (MA: Marketing Authorization ) en lugar de la Condicional (CMA: Conditional Marketing authorization)! Nota – la CMA fue emitida, pero las condiciones nunca fueron cumplidas por Pfizer/BioNTech, porque a quién le importa, era un juego desde el principio.
«(Co)-Rapps» = co-ponentes. La EMA es una entidad europea, compuesta por las «autoridades competentes» de los Estados miembros, que antes regulaban y aprobaban los productos farmacéuticos en cada país por separado. En la estructura europea, el equipo de revisión técnica y de co-revisión se selecciona para un producto específico. En el caso de las «vacunas» covid, el equipo sueco dirigido por Philip Josephson fue el ponente (revisor principal) y el equipo francés dirigido por Jean-Michel Race, el co-ponente. «CHMP» = Comité de Medicamentos de Uso Humano (en la EMA).

El correo electrónico está dirigido a Olga Solomon en la CE (Comisión Europea), y el jefe de Noel, Emer Cooke – Director Ejecutivo de la EMA y ex alto ejecutivo de la OMS está copiado. Aquí está Emer Cooke:

El pacto inteligente de Úrsula.
¿Te acuerdas de ella?

Ursula vonden Leyen – Comisaria de la UE, cuyos logros incluyen la negociación de increíbles contratos de suministro predatorios de Pfizer en nombre de todos los Estados miembros de la UE por mensajes de texto con el CEO de Pfizer Albert Bourla. En estos contratos, los países de la UE tuvieron que poner activos estatales como garantía, renunciar a todas las leyes de control de calidad, importación y protección del consumidor y renunciar a la soberanía nacional, es decir, no se les permitió cambiar la legislación con respecto a la responsabilidad de las vacunas por parte de sus propios parlamentos? Los contratos predatorios fueron completamente redactados para proteger los llamados «intereses comerciales de Pfizer». El siguiente intercambio de correos electrónicos está relacionado con los valientes esfuerzos de Ursula:

Hay un montón de acrónimos utilizados, los más relevantes son «CE» = Comisión Europea, «EM» = Estados miembros, «PE»=Parlamento Europeo. La frase clave es que Ursula está «dispuesta a llamar personalmente a los ministros de Sanidad competentes para evitar el uso del apartado 2 del artículo 5». ¿De qué se trata? El artículo 5(2) se refiere al «artículo 5(2) de la Directiva 2001/83» – autorización de uso de emergencia en un Estado miembro europeo, otorgada por cada uno de los Estados miembros por separado en sus propios países. La CMA es una autorización condicional de comercialización que expide la EMA para todos los miembros de la UE simultáneamente. La UE la anuncia como un proceso mucho más sólido que una EUA :
…la ACM sigue un marco controlado y sólido que ofrece salvaguardias que las autorizaciones de uso de emergencia no pueden ofrecer. En realidad, una autorización de uso de emergencia no es una autorización de la vacuna, sino una autorización del uso temporal de la vacuna no autorizada. La CMA garantiza que todas las obligaciones en materia de farmacovigilancia, controles de fabricación, incluidos los controles de lotes para las vacunas, y otras obligaciones posteriores a la autorización se apliquen de manera jurídicamente vinculante […]. En particular:
-Garantiza un seguimiento riguroso, a través del sistema de farmacovigilancia de la UE, de la seguridad del medicamento en toda la UE. […]
-Garantiza el seguimiento de la seguridad después de la autorización y permite la recogida de datos adicionales de forma estructurada. […].
-Lafabricación rigurosa, incluida la liberación de lotes para vacunas y la distribución, están sujetas a los mismos controles continuos que todos los medicamentos autorizados. La supervisión de los procesos de fabricación garantiza que el medicamento se fabrica y controla de acuerdo con normas farmacéuticas estrictas en el contexto de la comercialización a gran escala.
En virtud de una autorización condicional de comercialización (ACM) de la UE, la responsabilidad recae en el titular de la autorización de comercialización. El titular de la autorización de comercialización será responsable del producto y de su uso seguro.
En teoría suena muy bien. Es lo que prometió Úrsula cuando llamó personalmente a los políticos de los Estados miembros y les retorció el brazo. Tal vez ni siquiera fue necesario retorcerles el brazo, ya que estaban suficientemente aterrorizados por la propaganda del covid y esperando que las «vacunas» milagrosas les salvaran. El problema es que Úrsula nunca tuvo la intención de cumplir estas promesas y, en cualquier caso, no es posible producir las «vacunas» de ARNm con la seguridad, eficacia y calidad de fabricación exigidas a los productos farmacéuticos. Lo que Ursula realmente necesitaba de este proceso era atar a todos los Estados miembros europeos en un pacto prometiendo una AMC «robusta», para que no pudieran tener una autoridad independiente sobre las vacunas distribuidas en sus países. La vía del artículo 5 habría significado que cada EM podría autorizar el producto, y luego tendría la potestad de revocar la autorización si se detectara algún problema. El artículo 5 también prevé una exención de responsabilidad para el fabricante, pero imposibilita la autorización del producto. Con la vía de la CMA, ninguno de los Estados miembros podría ejercer la toma de decisiones independiente, por lo que ella podría obligarlos a todos a firmar los mismos contratos, insensatos y casi completamente redactados, de Pfizer, Moderna y AstraZeneca, que de todos modos eximían de toda responsabilidad, ¡y además prohibían a los países cambiar sus propias leyes con respecto a la responsabilidad!
Los compradores deben «indemnizar, defender y mantener indemne a Pfizer … de y contra cualquier y todos los juicios, reclamaciones, acciones, demandas, pérdidas, daños, responsabilidades, acuerdos, sanciones, multas, costes y gastos … que surjan de, en relación con, o como resultado de la Vacuna.»
Principales objeciones notables y ausencia de las mismas por parte de los revisores de la EMA.
La sección de Fabricación Química y Controles (CMC) de la Solicitud de Licencia Biológica es el pilar principal de la aprobación regulatoria. Describe el proceso de fabricación y el cumplimiento de las Buenas Prácticas de Fabricación (cGMP), y un amplio conjunto de leyes y reglamentos diseñados para garantizar la pureza, potencia, consistencia y seguridad de los medicamentos y productos biológicos producidos en masa. Los datos de seguridad y eficacia de los ensayos clínicos son inútiles si el fabricante no puede garantizar a las autoridades reguladoras y a la comunidad médica que 1) el producto en cuestión, según las especificaciones, se utilizó en los ensayos clínicos, 2) el producto se fabrica de forma consistente, es puro, de alta calidad, reproducible, con un proceso de fabricación y unos pasos de control bien caracterizados y predecibles, 3) se distribuirá comercialmente el mismo producto que se probó.
Se identificaron problemas con la sección CMC de la presentación de Pfizer:

Los evaluadores de CMC no estaban contentos con el plazo dado para la evaluación, ya que violaba todos los plazos normales incluso los urgentes, también, por un gran margen. Así que la solución fue que había que «presionar» a los evaluadores. Con ello sólo se conseguía un objetivo: forzar hasta la extenuación a las personas que podrían haber planteado problemas, para que simplemente se rindieran y siguieran adelante. Al fin y al cabo, los de arriba sabían muy bien que la revisión reglamentaria no tenía ningún sentido ni implicación en la falsa «aprobación», iba a ocurrir como fuera. En el Reino Unido, la MHRA ya admitió que no tenía delegación formal de autoridad para revisar y aprobar estas inyecciones, y estoy dispuesto a apostar que la EMA tampoco tenía tal autoridad.
Los evaluadores de bajo nivel del CMC no lo sabían, y estaban trabajando muy duro y muy probablemente de buena fe. A finales de noviembre, plantearon más de 140 objeciones formales a la presentación del CMC de Pfizer, que todavía tenía muchos problemas y faltaba información. Como referencia, entre 10 y 15 objeciones reglamentarias normalmente impiden que una solicitud farmacéutica siga adelante hasta que se resuelven las objeciones. Tres objeciones importantes, es decir, las banderas rojas formales se discuten específicamente en los correos electrónicos a continuación. Yo y otros escribimos extensamente sobre la MO#2 (falta de integridad del ARNm). Aquí hay un correo electrónico de uno de los revisores, Evdokia Korakianiti y una respuesta de Alexis Nolte discutiendo el problema y el impacto (completamente desconocido y potencialmente muy preocupante) sobre la eficacia y seguridad del producto:

El problema de la degradación del ARNm también fue debatido por los especialistas en Calidad CMC Ton van der Stappen, Experto Biofarmacéutico Senior de la Junta de Evaluación de Medicamentos (con sede en los Países Bajos), y Especialista en Calidad de la EMA:

y Brian Dooley, otro especialista en calidad farmacéutica de la EMA:

Las imágenes que aparecen a continuación proceden de la revisión de calidad presentada por ellos a la EMA. La primera imagen habla del problema, ya bien documentado, de la degradación del ARNm en diferentes lotes del producto de Pfizer. Aquí se enumeran los resultados de los análisis de lotes suministrados por Pfizer y se codifican por colores según los diferentes lugares de fabricación, así como por sustancia farmacológica y categoría de producto farmacológico. Sustancia farmacológica = componentes activos del producto (ARNm solo) y producto farmacológico es la sustancia formulada en los lípidos y otros ingredientes. El % de integridad del ARNm describe el % de ARNm de «longitud completa» detectado en un lote. la otra parte del lote estaba compuesta por piezas rotas desconocidas con propiedades o impacto en la seguridad desconocidos. Nótese que los reguladores no realizaron ninguna comprobación independiente de nada de esto, simplemente enumeraron las cifras proporcionadas por Pfizer/BioNTech.
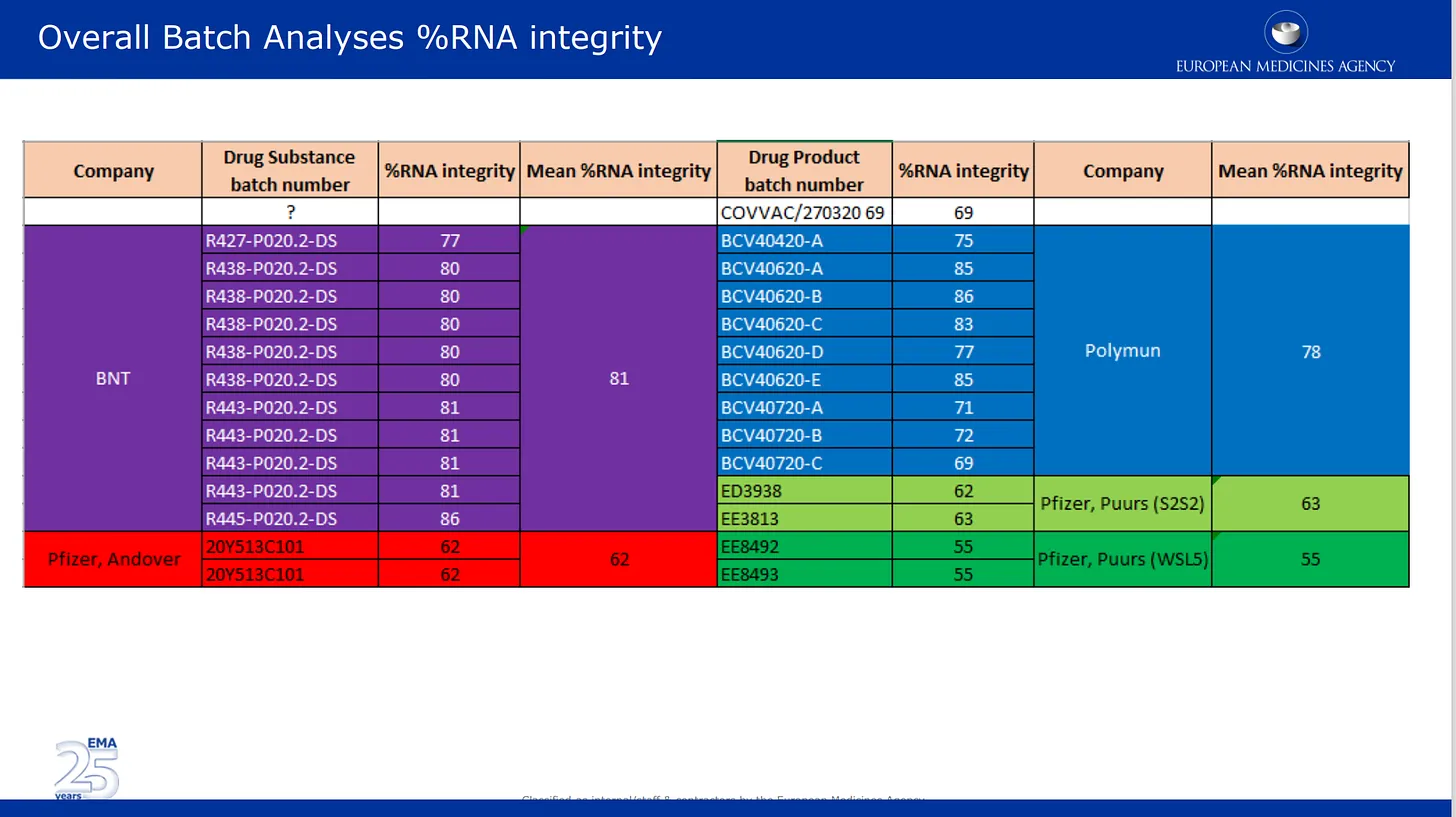
Parece que estos dos consultores científicos revisaron y aceptaron las imágenes falsas de los resultados presentados por Pfizer a la EMA – aquí están en su propia presentación PowerPoint de revisión del 24 de noviembre de 2020. Lea la nota debajo de la diapositiva – están aceptando estas imágenes como reales, a pesar de que estos dos revisores deberían saberlo mejor. ¿Por qué NO se opusieron a esto? Esto es lo que dice la nota:
El tamaño de la proteína después de la expresión in vitro de la sustancia farmacológica BNT162b2 se determinó mediante Western blot. Se confirmó que el tamaño de la proteína expresada era comparable para tres lotes del Proceso 1 y el lote del Proceso 2. La figura 3.2.S.2.6-15 muestra que el tamaño de la proteína expresada coincide con el tamaño esperado de la sustancia activa BNT162b2 y es comparable en todos los lotes analizados. Además, los niveles relativos de expresión son comparables en todos los lotes, como demuestra la intensidad comparable de las bandas en cada nivel de carga en todos los lotes.

Tal vez algunos periodistas contundentes deberían ponerse en contacto con los doctores van der Stappen y Dooley, y con la Sra. Korakianiti y otras personas mencionadas aquí para pedirles sus comentarios.
De las respuestas planteadas por Evdokia Korakianiti se desprende que la dirección de la EMA renunció a la pelea y se basó en «datos que sólo la FDA ha visto», pero es «optimista» y que la FDA afirmó que la rotura del ARNm era una «preocupación teórica». ¿De verdad? ¿Hay datos que apoyen esta afirmación o no? Aquí están los correos electrónicos que indican que las objeciones principales fueron escritas formalmente y posteriormente ignoradas por la EMA ya que el producto fue enviado comercialmente sólo un par de semanas más tarde. Las condiciones de la CMA nunca se cumplieron.
Esto confirma lo que ya sabemos: ni la EMA (ni la FDA, Health Canada, MHRA u otros reguladores) tenían ninguna autoridad real sobre estos productos o impacto sobre si iban a ser desplegados en el público . Todo fue un teatro de principio a fin.
Aquí están las tres objeciones principales que siguen sin resolverse hasta la fecha:

Y aquí tomando las afirmaciones de la FDA sin ningún cuestionamiento o evaluación formal de los datos por los reguladores de la EMA:

Lo que puedo decir para terminar – conté aproximadamente 70 personas diferentes mencionadas en el documento filtrado y en los correos electrónicos, que facilitaron esta trágica farsa – la «aprobación» del producto más letal jamás liberado en el mayor número de personas, lo que resulta en un número sin precedentes de muertos y heridos en todo el mundo. Quizás, con algunas excepciones, la mayoría de ellos fueron engañados en 2020 y no comprendieron que estaban participando en un crimen de guerra y firmando un fraude mortal. Creo que la mayoría de ellos ya lo saben, espero que estén suficientemente horrorizados por lo que han permitido, y espero que estas personas den un paso al frente como denunciantes y empiecen a hablar. Necesitamos respuestas.
Publicado originalmente por Due Diligence and Art
Sugerir una corrección