

Scrisori din lumea interlopă
Credit de imagine: Shutter Stock
Emailuri scurse de la EMA revizuite
La sfârșitul anului 2020, o colecție de documente – aproximativ 900 de pagini din secțiunea „vaccin” a Pfizer „Chemistry Manufacturing and Controls (CMC)” din cadrul cererii de reglementare adresate Agenției Europene pentru Medicamente (EMA) a fost scursă și trimisă mai multor jurnaliști. Documentele au inclus, de asemenea, schimburi de e-mailuri de la unii dintre evaluatori și de la cadrele superioare de la EMA. Scurgerea de informații a fost acoperită de British Medical Journal, care a reușit să confirme că documentele sunt autentice. Am primit aceste documente de la un coleg aproximativ un an mai târziu, la sfârșitul anului 2021, și am citit și utilizat o mare parte din ele în analizele mele. Am văzut multe documente legate de cercetare și dezvoltare de la Pfizer în activitatea mea profesională, așa că pot confirma, de asemenea, că aceste scurgeri de informații erau foarte conforme cu documentația tipică Pfizer. EMA nu a negat autenticitatea și a declarat doar că anteturile unora dintre e-mailuri au fost modificate.
Acum am un număr mult mai mare de adepți și o înțelegere mult mai clară a organizării și a structurii pseudo-juridice a cartelului criminal care conduce atrocitatea globală cunoscută colocvial sub numele de „răspunsul la pandemia de covid”. Revăd e-mailurile care au făcut obiectul unei scurgeri de informații, deoarece cred că acestea oferă dovezi extrem de importante.
Fișierele de e-mailuri EMA pe care le-am citit conțin 14 capturi de ecran ale unor e-mailuri de la mijlocul până la sfârșitul lunii noiembrie 2020. Schimburile provin de la personalul EMA și de la cadrele superioare. În opinia mea, aceste e-mailuri demonstrează că:
- Revizorii EMA au fost supuși unei presiuni politice masive pentru a inventa noi modalități de aprobare a produselor periculoase neaprobabile. Presiunea venea de la vârful guvernelor SUA, Regatului Unit și UE.
- Comisarul UE, Ursula von der Leyen, a făcut promisiuni statelor membre pe care nu intenționa să le îndeplinească niciodată pentru a le lega pe toate într-un singur pact pentru contractele de vaccinare și pentru a preîntâmpina astfel orice decizie independentă în țările lor.
- Au existat probleme grave și nerezolvabile – având în vedere calendarul intenționat nerealist – cu privire la calitatea produsului pe care personalul EMA a fost presat să îl aprobe. Unii nu s-au simțit confortabil să facă acest lucru și și-au exprimat îngrijorarea. Alții au „trecut cu vederea” date în mod clar inventate.
În cele din urmă, examinarea reglementară în sine și preocupările exprimate nu au contat – produsul urma să fie comercializat oricum. Acum știm exact de ce – autoritățile de reglementare nu au avut puterea de reglementare asupra acestuia. Autoritățile de reglementare în domeniul farmaceutic nu supraveghează materialele militare cunoscute sub numele de „contramăsuri” și „demonstrații de fabricație” (limbaj timid care acoperă agenții de război biologic produși de guvernul SUA capturat și de partenerii săi globali). E-mailurile arată că majoritatea personalului EMA au fost actori involuntari în această piesă de teatru.
Confirmarea acestui lucru pentru Marea Britanie a apărut recent.
Pe baza răspunsului la FOIA al MHRA:
„Toate deciziile de autorizare a vaccinurilor și a produselor terapeutice Covid au fost luate de către ministrul însărcinat cu acordarea licențelor și nu au fost delegate”
Traducere – în mod normal, autoritatea de a revizui și aproba noi produse farmaceutice este delegată în mod oficial MHRA de către secretarul de stat pentru sănătate (Marea Britanie). În cazul produselor covidiene, această delegare de autoritate nu există. Se pare că toate acestea au fost desfășurate de unul singur de către Matt Hancock (deși acesta arată cu degetul spre cineva mai sus). Același lucru s-a întâmplat și în SUA – Alex Azar, sub administrația Trump, a desfășurat aceste biomateriale neconforme pe americani, iar Xavier Becerra, sub administrația Biden, continuă să facă acest lucru și în prezent.
Presiunea politică.
Următorul schimb de e-mail a avut loc la 16 noiembrie 2020 între cadrele superioare ale EMA:
Noel Wathion – director executiv adjunct (pensionat în iunie 2021):

Agnes Saint-Raymond – șefa diviziei de afaceri internaționale:

Marco Cavaleri, președinte al Grupului operativ pentru pandemie Covid-19 din cadrul EMA:

E-mailurile trebuie citite de jos în sus.

Câteva lucruri interesante: cele trei autorități de reglementare – FDA din SUA, MHRA din Marea Britanie și EMA din UE – sunt toate ocupate să coordoneze momentul aprobării, înainte de a avea loc o analiză oficială a datelor, înainte ca comitetele consultative să fi văzut rezultatele studiilor clinice, să le fi discutat, să le fi votat etc. Aceștia discută despre momentul în care datele NU CONTEAZĂ în ceea ce privește introducerea sau nu pe piață a acestor produse. În plus, acestea interacționează ca și cum nu ar fi trei agenții separate ale unor națiuni suverane separate, responsabile față de seturi separate de contribuabili și de supravegherea congresuală/parlamentară, ci pur și simplu departamente birocratice deja fuzionate într-un singur guvern global. În cele din urmă, FDA se va „grăbi să intre în EUA”, fiind „împinsă de Azar” (Alex Azar – secretar HHS la acea vreme) și „Trump trage sforile”.
Mulți oameni mă întreabă cum este posibil ca mii de oameni să fi participat la escrocheria orchestrată sub numele de „răspuns la pandemia de covid” – cu siguranță nu este posibil să fie atât de mulți oameni în cârdășie! Nu era necesar să fie atât de mulți la curent. În acest caz, Noel Wathion, un cadru de top al EMA, fie nu știe că revizuirea datelor este irelevantă pentru ca injecțiile să fie în cele din urmă introduse pe piață, fie denaturează abil acest lucru (de fapt, cred că nu era conștient). Prin urmare, personalul EMA de sub el nu ar trebui să fie conștient, fiind pur și simplu grăbit să îndeplinească orice sarcină îngustă care le-a fost atribuită. Compartimentarea este cheia pentru a acoperi orice escrocherie majoră în cadrul organizațiilor mari și al structurilor complexe. Acesta este motivul pentru care a demisionat/retractându-se la scurt timp după lansarea loviturilor ucigașe? De asemenea, el se află sub presiunea CE (Comisia Europeană) pentru a aproba. Iar Pfizer vrea acum o Autorizație de punere pe piață (AMM) completă în loc de cea condiționată (AMC)! Notă – CMA a fost emisă, dar condițiile nu au fost niciodată îndeplinite de Pfizer/BioNTech, pentru că, cui îi pasă, a fost un joc de la început.
„(Co)-Rapps” = co-raportori. EMA este o entitate europeană, compusă din fostele „autorități competente” separate ale statelor membre, care obișnuiau să reglementeze și să aprobe produsele farmaceutice în fiecare țară în parte. În structura europeană, echipa de revizuire tehnică și de co-revizuire este selectată pentru un anumit produs. În cazul „vaccinurilor” covid, echipa suedeză condusă de Philip Josephson a fost raportor (examinator principal), iar echipa franceză condusă de Jean-Michel Race – coraportor. „CHMP” =Comitetul pentru medicamente de uz uman (în cadrul EMA).

E-mailul este adresat Olgăi Solomon de la CE (Comisia Europeană), iar șeful lui Noel, Emer Cooke – director executiv al EMA și fost cadru superior la OMS – este copiat. Iată-l pe Emer Cooke:

Pactul inteligent al Ursulei.
Vă mai amintiți de ea?

Ursula von der Leyen – comisar european, ale cărei realizări includ negocierea unor incredibile contracte de furnizare prădătoare ale Pfizer în numele tuturor statelor membre ale UE prin mesaje text cu directorul executiv al Pfizer, Albert Bourla. În aceste contracte, țările UE trebuiau să pună ca garanție active de stat, să renunțe la toate legile privind controlul calității, importul și protecția consumatorilor și să renunțe la suveranitatea națională – adică să nu li se permită modificarea legislației cu privire la răspunderea pentru vaccinuri de către propriile parlamente? Contractele prădătoare care au fost complet redactate pentru a proteja așa-numitele „interese comerciale ale Pfizer”. Următorul schimb de e-mail se referă la eforturile curajoase ale Ursulei:

Sunt folosite o grămadă de acronime, cele mai relevante fiind „EC” = Comisia Europeană, „MS” = statele membre, „EP”= Parlamentul European. Fraza cheie este că Ursula este „pregătită să cheme personal miniștrii sănătății relevanți pentru a evita utilizarea articolului 5 alineatul (2)” . Despre ce este vorba? Articolul 5 alineatul (2) se referă la „articolul 5 alineatul (2) din Directiva 2001/83” – autorizația de utilizare de urgență într-un stat membru european, acordată de fiecare stat membru în parte în țara sa. CMA este o autorizație de punere pe piață condiționată, care este emisă de EMA pentru toți membrii UE simultan. Anunțată de UE ca fiind un proces mult mai robust decât o EUA (subliniere adăugată de mine):
…CMA urmează un cadru controlat și robust care oferă garanții pe care autorizațiile de utilizare de urgență ar putea să nu le ofere. În realitate, o autorizație de utilizare de urgență nu este o autorizare a vaccinului, ci o autorizare a utilizării temporare a vaccinului neautorizat. AMC se asigură că toate controalele de farmacovigilență, controalele de fabricație, inclusiv controalele loturilor pentru vaccinuri și alte obligațiilor post-aprobare se aplică într-un mod obligatoriu din punct de vedere juridic […]. În special:
-Asigură o monitorizare riguroasă, prin intermediul sistemului de farmacovigilență al UE, a siguranței medicamentului în întreaga UE. […]
-Asigură monitorizarea siguranței post-autorizare și permite colectarea de date suplimentare într-un mod structurat. […].
-Fabricarea riguroasă,inclusiv eliberarea loturilor pentru vaccinuri și distribuția, sunt supuse acelorași controale permanente ca și în cazul tuturor medicamentelor autorizate. Monitorizarea proceselor de fabricație asigură faptul că medicamentul este fabricat și controlat în conformitate cu standarde farmaceutice ridicate în contextul comercializării pe scară largă.
În cadrul unei autorizații de introducere pe piață condiționată (CMA) a UE, răspunderea revine titularului autorizației de introducere pe piață. Titularul autorizației de introducere pe piață va fi responsabil pentru produs și pentru utilizarea în siguranță a acestuia.
În teorie, acest lucru sună grozav. Este ceea ce a promis Ursula atunci când a sunat personal și a răsucit brațele politicienilor din statele membre. Poate că nici măcar nu a fost necesară această răsucire de brațe, deoarece aceștia erau suficient de terorizați de propaganda Covid și așteptau ca „vaccinurile” miraculoase să îi salveze. Problema este că Ursula nu a avut niciodată intenția de a îndeplini aceste promisiuni și, în orice caz, nu este posibil să producă „vaccinurile” cu ARNm la nivelul de siguranță, eficacitate și calitate de fabricație cerut de produsele farmaceutice. Ceea ce Ursula a vrut cu adevărat din acest proces a fost să lege toate statele membre europene într-un pact prin promisiunea unei AMC „robuste”, astfel încât acestea să nu poată avea o autoritate independentă asupra vaccinurilor distribuite în țările lor. Calea articolului 5 ar fi însemnat că fiecare stat membru ar fi putut autoriza produsul, iar apoi ar fi avut puterea de a revoca autorizația în cazul în care ar fi fost detectate probleme. Articolul 5 prevede, de asemenea, o exonerare de răspundere a producătorului, însă face imposibilă mandatarea produsului. Cu calea CMA, niciunul dintre statele membre nu ar fi putut exercita un proces decizional independent, astfel încât aceasta ar fi putut să le forțeze pe toate să încheie aceleași contracte nebunești și aproape complet redactate de Pfizer, Moderna și AstraZeneca, care oricum renunțau la orice răspundere și, în plus, interziceau țărilor să își modifice propriile legi în ceea ce privește răspunderea!
Cumpărătorii trebuie „să despăgubească, să apere și să exonereze Pfizer … de orice proces, pretenții, acțiuni, cereri, pierderi, daune, responsabilități, înțelegeri, penalități, amenzi, costuri și cheltuieli … care decurg din, sunt legate de sau rezultă din vaccin”
Obiecții majore notabile și lipsa acestora din partea evaluatorilor EMA.
Secțiunea „Chemistry Manufacturing and Controls” (CMC) din cererea de autorizare a produselor biologice este principalul pilon al aprobării de reglementare. Aceasta descrie procesul de fabricație și conformitatea cu bunele practici de fabricație (cGMP), precum și cu un set extins de legi și reglementări menite să asigure puritatea, potența, consistența și siguranța medicamentelor și produselor biologice produse în masă. Datele privind siguranța și eficacitatea din studiile clinice sunt inutile dacă producătorul nu poate asigura autoritățile de reglementare și comunitatea medicală că: 1) produsul în cauză, conform specificațiilor, a fost utilizat în studiile clinice, 2) produsul este fabricat în mod consecvent, pur, de înaltă calitate, reproductibil, cu un proces de fabricație și etape de control bine caracterizate și previzibile, 3) același produs care a fost testat va fi distribuit comercial.
Au fost identificate probleme în secțiunea CMC din prezentarea Pfizer:

Evaluatorii CMC nu au fost mulțumiți de calendarul oferit pentru evaluare, deoarece acesta încălca toate termenele normale și, de asemenea, pe cele accelerate, cu o marjă mare. Astfel, soluția a fost că evaluatorii trebuiau doar „împinși”. Acest lucru a atins un singur scop – să forțeze persoanele care ar fi putut să ridice probleme până la limita epuizării, astfel încât acestea să renunțe pur și simplu și să se conformeze. La urma urmei, cei de la vârf știau foarte bine că evaluarea de reglementare nu avea nicio semnificație și nicio implicație asupra falsei „aprobări”, ci urma să se întâmple indiferent de situație. În Marea Britanie, MHRA a recunoscut deja că nu a avut nicio delegare formală de autoritate pentru a revizui și aproba aceste injecții și sunt gata să pariez că nici EMA nu avea o astfel de autoritate.
Evaluatorii CMC de nivel scăzut nu știau acest lucru și lucrau din greu și, cel mai probabil, cu bună credință. Până la sfârșitul lunii noiembrie, aceștia au ridicat peste 140 de obiecții formale la prezentarea CMC a Pfizer, care avea încă multe lacune și informații lipsă. Pentru referință, 10-15 obiecții de reglementare împiedică, în mod normal, ca o cerere farmaceutică să meargă mai departe până la soluționarea obiecțiilor. Trei obiecții majore, adică stegulețe roșii formale, sunt discutate în mod specific în e-mailurile de mai jos. Eu și alții am scris pe larg despre MO#2 (lipsa integrității ARNm). Iată un e-mail de la unul dintre evaluatori, Evdokia Korakianiti, și un răspuns de la Alexis Nolte care discută problema și impactul (complet necunoscut și potențial foarte îngrijorător) asupra eficacității și siguranței produsului:

Problema degradării ARNm a fost discutată, de asemenea, de către specialiștii în CMC Quality Ton van der Stappen, Senior Biopharmaceutical Expert la Medicines Evaluation Board (cu sediul în Olanda) și specialist în calitate pentru EMA:

și Brian Dooley, un alt specialist în calitate farmaceutică la EMA:

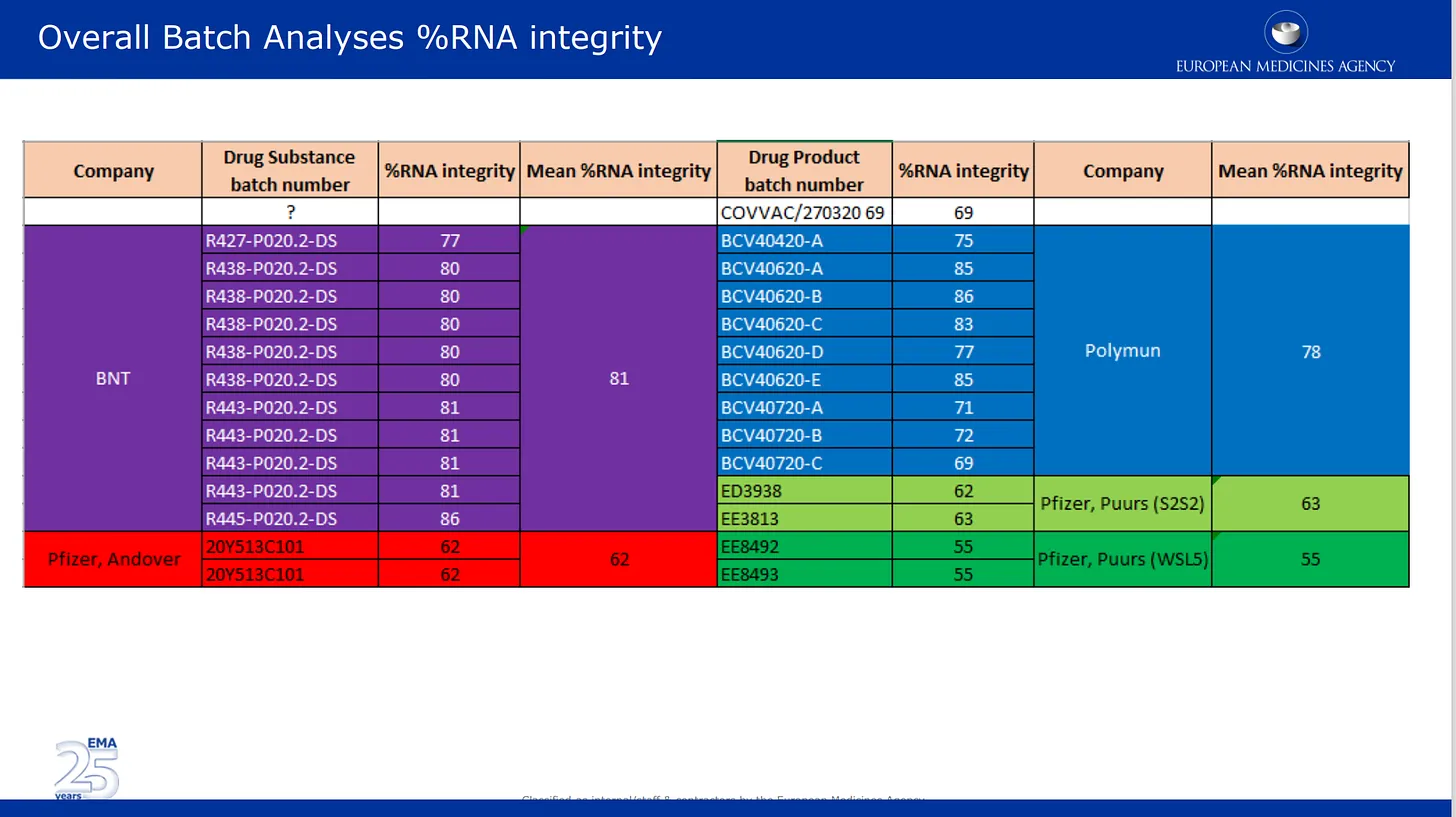
Imaginile de mai jos provin din analiza calității transmisă de aceștia către EMA. Prima imagine vorbește despre problema, acum bine documentată, a degradării ARNm în diferite loturi ale produsului Pfizer. Aici, rezultatele analizei loturilor furnizate de Pfizer sunt listate și codificate pe culori în funcție de diferitele locații de producție, precum și în funcție de substanța medicamentoasă și de categoria de produse medicamentoase. Substanța medicamentoasă = componentele active ale produsului (doar ARNm), iar produsul medicamentos este substanța formulată în lipide și alte ingrediente. Integritatea % ARNm descrie procentul de ARNm de „lungime completă” detectat într-un lot. cealaltă parte a lotului a fost compusă din bucăți rupte necunoscute cu proprietăți sau impact asupra siguranței necunoscute. Rețineți că autoritățile de reglementare nu au efectuat nicio verificare independentă a acestor date, ci doar au enumerat cifrele furnizate de Pfizer/BioNTech.
Separe că acești doi consultanți științifici au revizuit și au acceptat imaginile false ale rezultatelor Western blot transmise de Pfizer către EMA – iată-le în propria lor prezentare PowerPoint de revizuire din 24 noiembrie 2020. Citiți nota de sub diapozitiv – ei acceptă aceste imagini ca fiind reale, chiar dacă ambii recenzenți ar trebui să știe mai bine. De ce NU au obiectat la acest lucru? Iată ce scrie în notă:
Dimensiunea proteinei după exprimarea in vitro a substanței medicamentoase BNT162b2 a fost determinată cu ajutorul Western blot. Dimensiunea proteinelor exprimate a fost confirmată ca fiind comparabilă pentru cele trei loturi Proces 1 și lotul Proces 2. Figura 3.2.S.2.2.6-15 arată că dimensiunea proteinei exprimate este în concordanță cu dimensiunea așteptată a substanței medicamentoase BNT162b2 și comparabilă pentru toate loturile testate. În plus, nivelurile relative de expresie sunt comparabile pentru toate loturile, după cum reiese din intensitatea comparabilă a benzilor la fiecare nivel de încărcare în toate loturile.

Poate că niște jurnaliști incisivi ar trebui să contacteze doctorii van der Stappen și Dooley, precum și pe doamna Korakianiti și alte persoane menționate aici pentru a face comentarii.
Este evident din răspunsurile ridicate de Evdokia Korakianiti că managementul EMA a renunțat la arme și s-a bazat pe „date pe care doar FDA le-a văzut”, dar este „optimist” și că FDA a susținut că ruperea ARNm a fost o „preocupare teoretică”. Serios? Există date care să susțină această afirmație sau nu? Iată e-mailurile care indică faptul că obiecțiile majore au fost redactate în mod oficial și, ulterior, ignorate de către EMA, deoarece produsul a fost livrat în scop comercial doar câteva săptămâni mai târziu. Condițiile AMC nu au fost niciodată îndeplinite.
Acest lucru confirmă ceea ce știm deja – nici EMA (nici FDA, nici Health Canada, MHRA sau alte autorități de reglementare) nu au avut vreo autoritate reală asupra acestor produse sau impact asupra faptului că acestea urmau să fie utilizate asupra publicului neștiutor. Totul a fost un teatru de la început până la sfârșit.
Iată cele trei obiecții majore care au rămas nerezolvate până în prezent:

Și iată și niște fluturași de mână și preluarea afirmațiilor FDA fără nicio întrebare sau evaluare formală a datelor de către autoritățile de reglementare EMA:

Ceea ce pot spune în încheiere – am numărat aproximativ 70 de persoane diferite menționate în documentul și în e-mailurile care au făcut obiectul unor scurgeri de informații, care au facilitat această șaradă tragică – „aprobarea” celui mai letal produs lansat vreodată asupra celui mai mare număr de oameni, ceea ce a dus la un număr fără precedent de morți și răniți în întreaga lume. Poate că, cu câteva excepții, cei mai mulți dintre ei au fost înșelați în 2020 și nu au înțeles că participau la o crimă de război și că au semnat o fraudă mortală. Cred că cei mai mulți dintre ei știu până acum, sper că sunt suficient de îngroziți de ceea ce au permis și sper ca acești oameni să facă un pas înainte ca denunțători și să înceapă să vorbească. Avem nevoie de răspunsuri.
Publicat inițial de Due Diligence and Art
Suggest a correction




