Lo que revelan los correos electrónicos y documentos filtrados de la EMA: Principales preocupaciones con la integridad del lote de la vacuna C-19 de Pfizer y la carrera para autorizar
Nuestro artículo reciente de Meryl Nass, MD, sobre el lanzamiento de vacunas más rápido en la historia mundial, que detalla el procedimiento de aprobación de las nuevas «vacunas» de ARNm, destaca la ignorancia incomprensible de cómo funcionan las autoridades sanitarias en la actualidad.
Al mismo tiempo, el Comité Especial del Parlamento Europeo sobre la pandemia de COVID-19 tuvo la oportunidad de plantear preguntas a los representantes de cuatro compañías farmacéuticas el 5 de septiembre de 2022. La reunión se centró en las actividades pasadas y futuras relacionadas con la pandemia con un intento desesperado. por unos pocos para lograr la transparencia y la rendición de cuentas esperadas en un entorno democrático. El Sr. Cristian Terhes, miembro del Parlamento Europeo, ha planteado las preguntas correctas.
Para cualquiera que pueda estar sorprendido por el rápido lanzamiento de las nuevas ‘vacunas’ en Europa, debemos recordar y cuestionar cómo fue la autorización de la política de vacunación anterior COVID-19, como supimos de los correos electrónicos filtrados de la EMA (Agencia Europea de Medicamentos). ), resumido en el siguiente artículo de la periodista de investigación y locutora Sonia Elijah, en Trial Site News (junio de 2022).
Lo que revelan los correos electrónicos y documentos filtrados de la EMA: Principales preocupaciones con la integridad del lote de la vacuna C-19 de Pfizer y la carrera para autorizar
Por Sonia Elijah
Trial Site News ( video arriba ) pudo revisar recientemente los correos electrónicos internos filtrados de la Agencia Europea de Medicamentos (EMA) y el informe de la reunión entre la agencia y Pfizer. La EMA supervisa la evaluación y supervisión de medicamentos para la Unión Europea. Al igual que otros organismos reguladores de la salud, su principal responsabilidad es proteger y promover la salud pública. Instantáneas de la correspondencia interna por correo electrónico de la EMA; una presentación de PowerPoint del 26 de noviembre de 2020 de una reunión fundamental entre Pfizer y la agencia, así como un informe confidencial de Pfizer de 43 páginas fueron proporcionados por una fuente anónima debido a su confianza en el compromiso con la transparencia de Trial Site, la accesibilidad y la responsabilidad en la promoción de una industria de investigación biomédica altamente ética, centrada en la calidad y centrada en la salud pública.
Las agencias reguladoras, como la EMA, la Administración de Alimentos y Medicamentos (FDA) en los EE. UU. y la Agencia Reguladora de Medicamentos y Productos para el Cuidado de la Salud (MHRA) del Reino Unido, están facultadas para tomar decisiones basadas en mejorar al público. Sin embargo, las influencias externas, como la presión política o de los medios, no deben ser un factor determinante en su toma de decisiones, cuando se trata de condiciones pandémicas y la autorización de comercialización condicional acelerada de las vacunas Covid-19 (particularmente para el mRNA- basadas en vacunas producidas por Pfizer-BioNTech y Moderna), parece que este último ganó el día.
El período de tiempo de la correspondencia por correo electrónico en cuestión se extiende del 10 al 25 de noviembre de 2020, solo unas semanas antes de que la EMA otorgara la CMA (autorización de comercialización condicional) para la vacuna Pfizer-BioNTech Covdid-19 el 21 de diciembre de 2020. La FDA otorgó la EUA (autorización de uso de emergencia) para esta vacuna el 11 de diciembre y la MHRA llegó primero a la línea de meta el 2 de diciembre. Aquí este autor usa el término «línea de meta», ya que los correos electrónicos revelan una intensa, casi competitiva prisa por autorizar las vacunas contra el Covid-19, lo más rápido posible. Comprensiblemente, el mundo se vio afectado por una pandemia en ese momento, donde hubo un gran impulso para autorizar una vacuna para proteger a las personas del nuevo coronavirus.
La carrera hacia EUA
En un correo electrónico de Marco Cavaleri, en ese momento Jefe de Amenazas Biológicas para la Salud y Estrategia de Vacunas de la EMA, comunicó con urgencia cómo la FDA de los EE. UU.“va a precipitarse en EUA”.
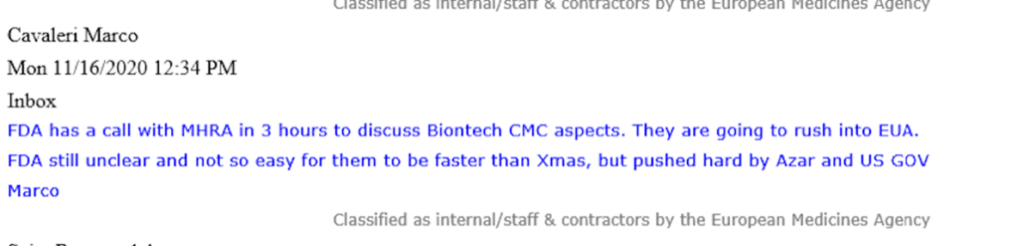
Cavaleri se refiere a que esta “aceleración” está siendo “impulsada con fuerza por Azar y el gobierno de EE. UU”. Bajo la administración de Trump, Alex Azar, exejecutivo farmacéutico, fue Secretario de Salud y Servicios Humanos de los Estados Unidos (HHS) de 2018 a 2021. La FDA es una agencia que cae directamente bajo el HHS.
Vale la pena señalar que cuando Azar era expresidente de Lilly USA LLC, una división de Eli Lilly, los precios de los medicamentos se dispararon bajo su liderazgo. La compañía farmacéutica también se vio envuelta en una demanda colectiva bajo su mandato en la que fue acusada de explotar el sistema de precios de medicamentos para aumentar las ganancias de su medicamento de insulina. Por supuesto, esto no significa necesariamente que este ejecutivo haya sido cómplice de alguna manera, pero la coordinación es notable.
El correo electrónico de Cavaleri habla de la medida en que la política (y el gobierno de los EE. UU.) estaban impulsando el proceso regulatorio de la FDA, asegurándose de que iba a “velocidad vertiginosa». Y, por supuesto, en esa nota, la Operación Warp Speed de Trump fue para garantizar que se rompieran todos los registros de desarrollo de vacunas. Sin duda, las intenciones eran buenas ante el estallido de la peor pandemia en un siglo.
Sin embargo, al otro lado del Atlántico, en Europa, la tensión de la agencia reguladora aumentó a medida que la presión para acelerar los plazos hizo que el ambiente y el estado de ánimo general se tensaran: la presión y la ansiedad eran palpables en los intercambios de correo electrónico revisados.
Surgieron personas de gran integridad y claridad en cuanto a sus funciones y compromisos como administradores de la salud pública. Por ejemplo, una persona demostró una preocupación palpable por los plazos acelerados para garantizar que cumplirían con el «plazo» para la autorización de la vacuna a expensas de una evaluación sólida. Era Noel Wathion, en ese momento director ejecutivo adjunto de la EMA, pero que desde entonces se jubiló . Este funcionario de la EMA señaló de manera importante: «Estamos acelerando tanto como sea posible, pero también debemos asegurarnos de que nuestra evaluación científica sea lo más sólida posible». No olvidemos la responsabilidad/rendición de cuentas adjunta a la recomendación a la CE de otorgar una CMA.’

Wathion supone que la EUA de la FDA (y la MHRA) se emitiría antes de que la EMA otorgara su propia CMA, lo que resultó ser correcto. Lo que es interesante es su preocupación por abordar la ‘limitación de daños‘ resultante del probable resultado de que la EMA termine última en esta carrera regulatoria y su temor de que esto resulte en que la opinión pública y los medios se vuelvan en contra de la agencia. La velocidad aparentemente reemplazó las preocupaciones de calidad según una revisión cuidadosa de estos correos electrónicos.
En un correo electrónico del 19 de noviembre, Wathion revela una TC (llamada de teleconferencia) «bastante tensa» con la comisaria europea (Ursula von der Leyen) que fue «a veces incluso un poco desagradable«. Esto refleja la creciente presión a la que estaba sometido el personal de la EMA para emitir una CMA rápidamente después de una EUA otorgada por la FDA/MHRA para la vacuna Pfizer-BioNTech. Von der Leyen está implicada en ser potencialmente responsable de este ambiente tenso con ‘un retraso de varias semanas… no fácilmente aceptable para la CE [Comisión Europea]‘.
A principios de 2022, Trial Sites News informó cómo von der Leyen se vio envuelta en un escándalo cuando un grupo de eurodiputados independientes exigió su renuncia inmediata y la divulgación completa de una serie de mensajes de texto privados entre ella y el director ejecutivo de Pfizer, Albert Bourla. Solo una pequeña porción de estos textos fue alguna vez revelada. De los que sí lo fueron, ¡Revelaron que ella negoció partes de un acuerdo de vacunas en toda Europa, unilateralmente con Bourla a través de una serie de mensajes de texto! Claramente, los protocolos estándar en Europa se tiraron por la ventana a favor de la conveniencia y esto aparentemente estaba ligado a una presión competitiva unificada en las tres agencias reguladoras.
Wathion deja al descubierto sus reflexiones después de este TC en particular, y escribe sorprendentemente cómo » las consecuencias políticas parecen ser demasiado altas, incluso si el nivel «técnico» en los EM [Estados miembros] pudiera defender tal retraso para hacer que el resultado de la revisión científica lo más sólida posible“. Dicho de otra manera”, la difusión continua de ciencia apareció por primera vez como una tapadera de la política primero.

Wathion señala que un posible retraso de varias semanas para garantizar una «garantía sólida en particular con respecto a la CMC y la seguridad» se enfrentará a «críticas de varias partes«, incluidos los medios de comunicación, la CE (Comisión Europea) y el PE (Parlamento Europeo). Wathion habla de su temor de que si no se puede cumplir la fecha límite ‘para alinearse tanto como sea posible con el tiempo de «aprobación» de la FDA/MHRA’, nos veremos abrumados por todos los frentes y estaremos en medio de la tormenta”. Sin embargo, este posible retraso parecía ser necesario «para que el resultado de la revisión científica fuera lo más sólido posible«. Esto implica que la velocidad a expensas de la seguridad estaba a la orden del día para evitar las «consecuencias políticas«. Claramente, la política dictaba el protocolo de autorización de la vacuna Covid-19, no la ciencia.

En el correo electrónico anterior de Marco, el funcionario de la EMA revela que el director ejecutivo de Pfizer, Albert Bourla, presionó a Peter Marks, y esto podría interpretarse como muy controvertido, dado que Marks es el director del Centro de Evaluación e Investigación Biológica (CBER) de la FDA. El aparente acceso de Pfizer al organismo de control federal plantea al menos preguntas importantes, si no introduce la posibilidad de enredos inquietantes entre la industria y una agencia federal científica supuestamente independiente.
Principales preocupaciones con la integridad entre lotes de vacunas
Un correo electrónico de Cavaleri (ver más abajo) revela que en ese momento la FDA sabía de ‘algunos problemas‘ asociados con el CMC que debían resolverse y que pueden ‘terminar siendo la parte difícil‘. CMC se refiere a Química, Fabricación y Controles, también conocida como calidad farmacéutica, que cubre varios procedimientos utilizados para evaluar y garantizar la seguridad y consistencia entre lotes de productos farmacéuticos.
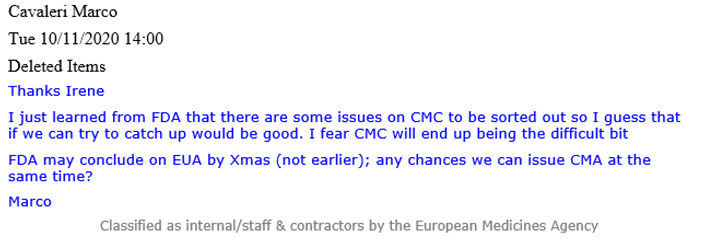
Un correo electrónico de Evdokia Korakianiti (administrador científico de la EMA) explica con más detalle cuáles eran estos «problemas» y cómo eran, de hecho, las principales preocupaciones relacionadas con la vacuna Pfizer-BioNTech.

De manera alarmante, se observaron diferencias significativas en los niveles de integridad del ARNm entre los lotes de vacunas comerciales (a gran escala) y clínicas (a pequeña escala) de Pfizer-BioNTech. ‘ ~78% de integridad del ARNm‘ en los clínicos y ‘~ 55% en los lotes comerciales propuestos‘ con la ‘causa raíz‘ aún no identificada. Las implicaciones de seguridad y eficacia debidas a esta preocupación también se señalaron en el correo electrónico «aún por definir«.
En un informe confidencial de Pfizer, que también se filtró junto con los correos electrónicos de la EMA, la empresa afirma que, según la experiencia general propia de Acuitas Therapeutics (la empresa de biotecnología que desarrolló la plataforma de nanopartículas lipídicas para la vacuna de Pfizer y Moderna), «un umbral mínimo es aproximadamente el 70%.‘ (Ver captura de pantalla a continuación)

Luego, en la página 30, dice: ‘La eficacia del producto depende de la expresión del ARN entregado, que requiere una molécula de ARN suficientemente intacta’. (Ver captura de pantalla a continuación)

Esta frase exacta «requiere una molécula de ARN suficientemente intacta» se usó en el correo electrónico del miembro del personal de EMA, Evdokia Korakianiti, que incluí anteriormente, enviado el 23 de noviembre de 2020; ahora probablemente sabemos de dónde lo hizo referencia Korakianiti.
Que los lotes comerciales (que iban a distribuirse por todo el mundo) tengan un nivel significativamente más bajo de integridad de ARNm (molécula de ARN intacta) es muy preocupante dado su vínculo intrínseco con la eficacia y la seguridad potencial del producto.
Al día siguiente, Veronika Jekerle, Jefa de la Oficina de Calidad Farmacéutica, escribe a Evdokia (ver más abajo).

La diferencia en el nivel de integridad del ARNm se señaló nuevamente como una preocupación importante » compartida por la mayoría de los estados miembros» y su » impacto potencial en la seguridad». Jekerle destaca en negrita: «Una aprobación para fin de año podría ser posible, si estas preocupaciones + GMP se resuelven«.
Esto da lugar a la pregunta crítica: ¿cómo se resolvieron todas estas preocupaciones cuando se otorgó CMA solo unas semanas después, el 21 de diciembre? Una posible forma en que se resolvió se explica más adelante en este informe.
En contraste con las preocupaciones de algunos de los otros funcionarios de la EMA, Marco Cavaleri escribe casi al mismo tiempo en el siguiente correo electrónico (ver más abajo) que el contenido de ARNm no es una preocupación importante, según la FDA –“la cuestión sobre el contenido de ARNm no se percibe como importante.’ También afirma sorprendentemente, «no está claro si alguna vez se realizaron inspecciones de GCP”. Esta revelación es muy preocupante dado que GCP se refiere a Buenas Prácticas Clínicas, que es ‘un estándar internacional de calidad ética y científica para diseñar, realizar, registrar e informar ensayos que involucran la participación de sujetos humanos’.
Lo que es aún más alarmante es su siguiente declaración: ‘No hay mayor interés por parte de la FDA‘. Esto parece revelar la aparente falta de preocupación o incluso de interés de la agencia reguladora sobre si se completaron las inspecciones de GCP, en el contexto de los ensayos clínicos de Pfizer, en los que la FDA confió para otorgar EUA para la vacuna Pfizer-BioNTech. En uno de los informes de investigación anteriores de este autor para Trial Site News , notamos que la FDA solo inspeccionó el 1% de los sitios de prueba de Pfizer.

Se revela más información condenatoria (ver captura de pantalla abajo) cuando múltiples agencias reguladoras: Health Canada (HC), EMA, MHRA y FDA son conscientes del problema de la integridad del % de ARNm, pero la FDA y Health Canada afirman sin fundamento que «las preocupaciones de seguridad asociadas. Son más bien una preocupación teórica«.

A continuación, Health Canada parece contradecirse, ya que más tarde se describe como una preocupación especial por el hecho de que una región reciba «todo el material subóptimo«. Obviamente, no quería ser esa región.
Sorprendentemente, el final del correo electrónico revela que el ‘Solicitante [Pfizer] ha compartido con la FDA y nosotros [EMA]/MHRA solo hoy y el problema con partículas visibles en el DP [producto farmacéutico] parece ser componentes de nanopartículas lipídicas).’
Esto es muy preocupante debido a que este importante problema se dio a conocer a las tres agencias reguladoras clave el 25 de noviembre, solo unas pocas semanas antes de que la EMA otorgara CMA y la FDA otorgara EUA para la vacuna de Pfizer. De manera alarmante, solo pasaron unos días antes de que la MHRA otorgara la autorización en el Reino Unido el 2 de diciembre de 2020. La suposición de Veronika de que las » partículas visibles» podrían ser LNP (nanopartículas lipídicas) es difícil de aceptar dado que las nanopartículas no son visibles a simple vista. Se observaron otras anomalías, aunque probablemente se trataba de un esfuerzo histórico en términos de velocidad de desarrollo de vacunas. Parece claro, sin embargo, se necesitaba algo más de tiempo.
Cómo se resolvió aparentemente el % de integridad del ARNm
La discrepancia entre los lotes parece haber sido resuelta cuando se menciona que los ‘últimos lotes [recibidos por la FDA] indican que el % de ARN intacto está alrededor del 70-75%’.
Sin embargo, en un informe filtrado de una reunión con Pfizer y la EMA el 26 de noviembre de 2020, un día después del correo electrónico de Veronika, sorprendentemente revela que la especificación de integridad del ARN se revisó a >=50 % para la vida útil del producto farmacéutico, significativamente menor que el umbral mínimo del 70% que había estipulado Acuitas Therapeutics y la media del 78% de los lotes clínicos. ¿Era esta la forma de la EMA (y potencialmente de la FDA/MHRA/HC) de ‘resolver’ el problema para garantizar ‘una aprobación para fin de año’?

Se hace mención a las ‘ incertidumbres sobre la consistencia de la calidad del producto y, por lo tanto, la incertidumbre en cuanto a la seguridad del producto y la eficacia del producto comercial’. Sin embargo, es desconcertante cómo la reducción de la especificación de integridad del ARN podría remediar esa gran objeción.
En otra diapositiva, el artefacto afirma: «Las especies de ARN truncado [acortado] y modificado deben considerarse impurezas relacionadas con el producto». Esto confirma que estas especies de mRNA acortadas que redujeron el nivel de integridad del %mRNA se clasificaron como impurezas. Otra preocupación alarmante que surge de estas impurezas se marca «debe abordarse la posibilidad de que se traduzcan proteínas distintas de la proteína pico prevista (S1 S2) como resultado de especies de ARNm truncadas y/o modificadas». (Ver captura de pantalla a continuación)

La evidencia en este informe confirma que los organismos reguladores como la FDA, MHRA, EMA y Health Canada conocían las diferencias en los lotes, con respecto al % de integridad del ARNm y, por lo tanto, se desconocía el efecto sobre la «seguridad y eficacia«. El informe filtrado de la reunión de Pfizer/EMA plantea preocupaciones importantes, suponiendo que el problema se resolvió simplemente reduciendo la especificación de integridad del ARN. En otras palabras, quizás nunca se resolvió.
Un sitio web en particular que ha llamado mucho la atención recientemente, que habla de la diferencia entre lotes es howbadismybatch.com . Es una base de datos integral con análisis sobre ‘código de lote y muertes, discapacidades y enfermedades asociadas para las vacunas Covid 19’. Al ingresar un número de lote de cualquiera de las vacunas Covid-19, le informa la frecuencia de los eventos adversos informados asociados con ese lote.
Hablé con Sasha Latypova, que ha realizado ensayos clínicos durante más de 25 años y es propietaria de su propia empresa de biotecnología, para pedirle su opinión experta sobre los documentos filtrados. Ella dijo:
“La falta de integridad del ARNm y la presencia de fragmentos de ARN no caracterizados en los lotes del producto de Pfizer se identificaron como una «Objeción importante» – una bandera roja reglamentaria formal, considerada una impureza del producto y habría sido un factor decisivo en cualquier proceso normal de aprobación de medicamentos. Como mínimo, requería un ensayo clínico «puente» adicional para evaluar los efectos clínicos que hubiera llevado meses diseñar y realizar correctamente. El pánico anuló la integridad científica y se adoptó un estándar de aceptación de lotes reducido arbitrariamente con el fin de cumplir con un plazo por motivos políticos. Hasta la fecha, este problema sigue sin resolverse y podría ser la causa subyacente de la enorme variación en las tasas de eventos adversos y muertes observadas para diferentes números de lote de fabricación en CDC VAERS y otras bases de datos”.
Latypova hizo una acertada referencia al destino del Titanic, al hacer una comparación en la forma en que los organismos reguladores llevaron a cabo su proceso de autorización de las vacunas contra el covid-19. El capitán del Titanic, Edward J. Smith, tenía como objetivo mejorar el tiempo de travesía de otro barco, lo que significaba que el barco viajaba demasiado rápido en aguas que se sabía que tenían hielo. Esto provocó una colisión fatal con un iceberg y el resto es historia.
A la luz de la evidencia incluida en este informe y el hecho de que la vacuna Pfizer-BioNTech Covid-19 es uno de los productos más lucrativos de la historia (el año pasado, Pfizer obtuvo 37 mil millones $ en ventas con predicciones para 2022 de $ 32 mil millones), este autor se esfuerza por abrir una discusión con algunas preguntas vitales que deben ser abordadas por las agencias reguladoras involucradas, Pfizer y aquellos en la comunidad científica/médica:
¿Cuáles son las implicaciones de seguridad y eficacia de una integridad de ARNm significativamente reducida (que surge de ARNm truncado y modificado) en los lotes comerciales de esta vacuna?
¿Cuáles son exactamente las partículas visibles observadas en el DP (producto farmacéutico) que Pfizer compartió en el último minuto con la EMA, FDA y MHRA y cuáles son sus implicaciones de seguridad y eficacia?
Las respuestas a estas preguntas son de gran importancia pública.
Trial Site News ha podido revisar recientemente los correos electrónicos internos filtrados de la Agencia Europea del Medicamento (EMA) y el informe de la reunión entre la agencia y Pfizer. La EMA supervisa la evaluación y la supervisión de los medicamentos para la Unión Europea. Al igual que otros organismos sanitarios reguladores, su principal responsabilidad es proteger y promover la salud pública. Una fuente anónima facilitó instantáneas de la correspondencia electrónica interna de la EMA; una presentación en PowerPoint del 26 de noviembre de 2020 de una reunión fundamental entre Pfizer y la agencia, así como un informe confidencial de 43 páginas de Pfizer, debido a su confianza en el compromiso de Trial Site con la transparencia, la accesibilidad y la responsabilidad en la promoción de una industria de investigación biomédica altamente ética, centrada en la calidad y en la salud pública.
Las agencias reguladoras, como la EMA, la Administración de Alimentos y Medicamentos (FDA) de Estados Unidos y la Agencia Reguladora de Medicamentos y Productos Sanitarios (MHRA) del Reino Unido, tienen la misión de tomar decisiones en beneficio del público. Las influencias externas, como la presión política o de los medios de comunicación, no deben ser un factor determinante en su toma de decisiones; sin embargo, cuando se trata de condiciones pandémicas y de la autorización de comercialización condicional por la vía rápida de las vacunas Covid-19 (en particular para las vacunas basadas en ARNm producidas por Pfizer-BioNTech y Moderna), parece que esto último ganó la partida.
Publicado originalmente en Trial Site News
Por favor, vea también: Mi presentación del informe de los correos electrónicos filtrados de la EMA al Dr. Reiner Fuellmich y su Comité de Investigación de Corona sobre Sonia Elijah Substack aquí.


![Una llamada a Europa: El futuro de nuestros hijos está en juego – Conf. de prensa histórica en Bruselas el 23 de enero [10 vídeos con subtítulos]](https://childrenshealthdefense.eu/wp-content/uploads/2022/01/chdeu-brussels-768x400.jpg)