Lettres de la pègre
Crédit photo : Shutter Stock
Les fuites de mails de l’EMA réexaminées
À la fin de l’année 2020, un ensemble de documents – environ 900 pages de la section CMC (Chemistry Manufacturing and Controls/Fabrication de produits chimiques et contrôles) du vaccin de Pfizer dans le cadre de la soumission réglementaire à l’Agence européenne des médicaments (EMA) – a fait l’objet d’une fuite et a été envoyé à un certain nombre de journalistes. Les documents comprenaient également des échanges de mails entre certains évaluateurs et des cadres supérieurs de l’EMA. La fuite a été reprise par le British Medical Journal, qui a pu confirmer que les documents étaient authentiques. J’ai reçu ces documents d’un collègue environ un an plus tard, à la fin de l’année 2021, et j’en ai lu et utilisé une grande partie dans mes analyses. J’ai vu de nombreux documents de Pfizer relatifs à la recherche et au développement dans le cadre de mon travail professionnel, et je peux donc confirmer que les documents qui ont fait l’objet d’une fuite sont tout à fait conformes à la documentation habituelle de Pfizer. L’EMA n’a pas nié l’authenticité de ces documents et a seulement déclaré que les en-têtes de certains mails avaient été modifiés.
J’ai maintenant beaucoup plus d’adeptes et une compréhension beaucoup plus claire de l’organisation et de la structure pseudo-juridique du cartel criminel à l’origine de l’atrocité mondiale familièrement appelée « réponse à la pandémie de covidies ». Je reviens sur les mails qui ont fait l’objet d’une fuite, car je pense qu’ils apportent des éléments de preuve extrêmement significatifs.
Les fichiers de mails de l’EMA que j’ai lus contiennent 14 captures d’écran de mails datant de la mi-novembre à la fin novembre 2020. Les échanges proviennent du personnel et des cadres supérieurs de l’EMA. À mon avis, ces mails démontrent que:
- Les évaluateurs de l’EMA étaient soumis à une pression politique massive pour inventer de nouvelles façons d’approuver les produits dangereux non approuvables. Ces pressions émanaient des plus hautes sphères des gouvernements des États-Unis, du Royaume-Uni et de l’Union européenne.
- La commissaire européenne Ursula von der Leyen a fait aux États membres des promesses qu’elle n’a jamais eu l’intention de tenir afin de les lier tous dans un pacte unique pour les contrats de vaccins et d’empêcher ainsi toute décision indépendante dans leur propre pays.
- Le personnel de l’EMA a été contraint d’accepter des problèmes graves et insolubles – compte tenu du calendrier volontairement irréaliste – en ce qui concerne la qualité du produit. Certains n’étaient pas à l’aise pour le faire et ont fait part de leurs préoccupations. D’autres ont « négligé » des données clairement inventées.
En fin de compte, l’examen réglementaire lui-même et les préoccupations soulevées n’ont pas eu d’importance – le produit allait être commercialisé de toute façon. Nous savons maintenant exactement pourquoi: les autorités réglementaires n’avaient pas de pouvoir réglementaire sur ce produit. Les organismes de réglementation pharmaceutique ne supervisent pas les matériaux militaires connus sous le nom de « contre-mesures » et de « produit de test » (un langage trompeur qui dissimule les agents de guerre biologique fabriqués par le gouvernement américain capturé et ses partenaires mondiaux). Les mails montrent que la majorité du personnel de l’EMA était un acteur involontaire de cette pièce.
La confirmation de ce fait pour le Royaume-Uni est apparue récemment.
Sur la base de la réponse à la FOIA de la MHRA:
« Toutes les décisions relatives aux vaccins et aux autorisations thérapeutiques de Covid ont été prises par le ministre chargé des licences et n’ont pas été déléguées
Traduction : normalement, le pouvoir d’examiner et d’approuver de nouveaux produits pharmaceutiques est officiellement délégué à la MHRA par le secrétaire d’État à la santé (Royaume-Uni). Dans le cas des produits covidiques, la délégation de pouvoir n’existe pas. Il semble que toutes ces mesures aient été déployées par Matt Hancock seul (bien qu’il pointe du doigt quelqu’un de plus haut placé). La même chose s’est produite aux États-Unis – Alex Azar, sous l’administration Trump, a déployé ces biomatériaux non conformes sur des Américains, et Xavier Becerra, sous Biden, continue de le faire aujourd’hui.
La pression politique.
L’échange de mails suivant a eu lieu le 16 novembre 2020 entre des cadres supérieurs de l’EMA :
Noel Wathion – Directeur exécutif adjoint (parti à la retraite en juin 2021) :

Agnès Saint-Raymond – chef de la division des affaires internationales :

Marco Cavaleri, président de la task force pandémie Covid-19 à l’EMA :

Les mails doivent être lus de bas en haut.

Quelques points intéressants : les trois régulateurs – la FDA américaine, la MHRA britannique et l’EMA européenne – sont tous occupés à coordonner le calendrier d’ approbation avant tout examen formel des données, avant que les comités consultatifs aient pris connaissance des résultats des essais cliniques, les aient discutés, aient voté à leur sujet, etc. Ils discutent du calendrier alors que les données n’ont AUCUNE IMPORTANCE pour la mise sur le marché ou non de ces produits. En outre, ils interagissent comme s’ils n’étaient pas trois agences distinctes de nations souveraines distinctes responsables de groupes de contribuables distincts et d’un contrôle parlementaire/congrès distinct, mais simplement des départements bureaucratiques déjà fusionnés au sein d’un gouvernement mondial. Enfin, la FDA va « se précipiter dans l’EUA », étant « poussée par Azar » (Alex Azar – secrétaire au HHS à l’époque) et « Trump tire les ficelles ».
De nombreuses personnes me demandent comment il est possible que des milliers de personnes aient participé à l’escroquerie orchestrée sous le nom de « réponse à la pandémie de grippe » – il n’est certainement pas possible d’avoir autant de personnes de mèche! Il n’était pas nécessaire d’avoir autant de personnes au courant. Ici, Noel Wathion, un haut responsable de l’EMA, ne sait pas que l’examen des données n’a rien à voir avec la question de savoir si les injections seront finalement mises sur le marché, ou bien il déforme habilement cette information (je pense en fait qu’il n’était pas au courant). Par conséquent, le personnel de l’EMA en dessous de lui n’aurait pas besoin d’être au courant, et serait simplement pressé d’accomplir la tâche étroite qui lui a été assignée. Le cloisonnement est la clé de la dissimulation de toute escroquerie majeure au sein de grandes organisations et de structures complexes. Est-ce la raison pour laquelle il a démissionné/pris sa retraite peu de temps après la mise en œuvre des tirs de destruction? Il est également soumis à la pression de la CE (Commission européenne) pour approuver le produit. Et Pfizer veut maintenant une autorisation de mise sur le marché (AMM) complète au lieu d’une autorisation conditionnelle (CMA)! Remarque : l’AMM a été délivrée, mais les conditions n’ont jamais été remplies par Pfizer/BioNTech, parce qu’on s’en fiche, c’était un jeu depuis le début.
« (Co)-Rapps » = co-rapporteurs. L’EMA est une entité européenne composée des anciennes « autorités compétentes » des États membres, qui réglementaient et approuvaient les produits pharmaceutiques dans chaque pays séparément. Dans la structure européenne, l’équipe d’examen technique et de co-examen est sélectionnée pour un produit spécifique. Dans le cas des « vaccins » covidés, l’équipe suédoise dirigée par Philip Josephson était le rapporteur (examinateur principal) et l’équipe française dirigée par Jean-Michel Race – le co-rapporteur. « CHMP » = Comité des médicaments à usage humain (au sein de l’EMA).

Le mail est adressé à Olga Solomon à la CE (Commission européenne), et le patron de Noel, Emer Cooke – directeur exécutif de l’EMA et ancien cadre supérieur de l’OMS – est mis en copie. Voici Emer Cooke :

Le pacte intelligent d’Ursula.
Vous vous souvenez d’elle ?

Ursula von der Leyen, commissaire européenne, qui a notamment négocié par SMS avec Albert Bourla, PDG de Pfizer, d’incroyables contrats d’approvisionnement prédateurs pour le compte de tous les États membres de l’UE. Dans le cadre de ces contrats, les pays de l’UE ont dû mettre en garantie des actifs de l’État, renoncer à toutes les lois relatives au contrôle de la qualité, à l’importation et à la protection des consommateurs, et abandonner leur souveraineté nationale – c’est-à-dire qu’ils n’ont pas été autorisés à modifier la législation relative à la responsabilité en matière de vaccins par leurs propres parlements ? Les contrats prédateurs qui ont été complètement expurgés pour protéger les soi-disant « intérêts commerciaux de Pfizer ». L’échange de mails suivant concerne les efforts courageux d’Ursula :

De nombreux acronymes sont utilisés, les plus pertinents étant « EC » = Commission européenne, « MS » = États membres, « EP »=Parlement européen. La phrase clé est qu’Ursula est « prête à appeler personnellement les ministres de la santé concernés pour éviter le recours à l’article 5, paragraphe 2 » . De quoi s’agit-il ? L’article 5, paragraphe 2, fait référence à l’article 5, paragraphe 2, de la directive 2001/83. Il s’agit d’une autorisation d’utilisation d’urgence dans un État membre de l’Union européenne, accordée par chacun des États membres séparément dans leur propre pays. L’AMC est une autorisation conditionnelle de mise sur le marché délivrée par l’EMA pour tous les membres de l’UE simultanément. Elle est présentée par l’UE comme une procédure beaucoup plus solide qu’une AUE (c’est moi qui souligne) :
…l’AMC suit un cadre contrôlé et solide offrant des garanties que les autorisations d’utilisation d’urgence pourraient ne pas avoir. En réalité, une autorisation d’utilisation d’urgence n’est pas une autorisation du vaccin mais une autorisation de l’utilisation temporaire du vaccin non autorisé. L’AMC veille à ce que toutes les mesures de pharmacovigilance, les contrôles de fabrication, y compris les contrôles de lots pour les vaccins, et les autres obligations post-approbation s’appliquent de manière juridiquement contraignante […]. Notamment :
-Elle garantit un suivi rigoureux, par le biais du système de pharmacovigilance de l’UE, de la sécurité du médicament dans l’ensemble de l’UE. […]
-Il garantit une surveillance de la sécurité après l’autorisation et permet la collecte de données supplémentaires de manière structurée. […].
-La fabrication rigoureuse, y compris la libération des lots pour les vaccins et la distribution, est soumise aux mêmes contrôles continus que pour tous les médicaments autorisés. La surveillance des processus de fabrication garantit que le médicament est fabriqué et contrôlé selon des normes pharmaceutiques élevées dans le contexte d’une commercialisation à grande échelle.
Dans le cadre d’une autorisation de mise sur le marché conditionnelle (AMC) de l’UE, la responsabilité incombe au titulaire de l’autorisation de mise sur le marché. Le titulaire de l’autorisation de mise sur le marché sera responsable du produit et de son utilisation en toute sécurité.
En théorie, c’est formidable. C’est ce qu’Ursula a promis lorsqu’elle a personnellement appelé les responsables politiques des États membres pour leur tordre le bras. Il n’était peut-être même pas nécessaire de leur tordre le bras, car ils étaient suffisamment terrorisés par la propagande covide et attendaient les « vaccins » miracles pour les sauver. Le problème est qu’Ursula n’a jamais eu l’intention de tenir ces promesses et que, de toute façon, il n’est pas possible de produire les « vaccins » à ARNm avec la sécurité, l’efficacité et la qualité de fabrication requises pour les produits pharmaceutiques. Ce qu’Ursula attendait vraiment de ce processus, c’était de lier tous les États membres de l’Union européenne dans un pacte en promettant une AMC « solide », afin qu’ils ne puissent pas avoir d’autorité indépendante sur les vaccins distribués dans leur pays. La voie de l’article 5 aurait permis à chaque État membre d’autoriser le produit et d’avoir le pouvoir de révoquer l’autorisation en cas de problème. L’article 5 prévoit également une exonération de responsabilité pour le fabricant, mais il est impossible de rendre le produit obligatoire. Avec la voie de la CMA, aucun des États membres ne pourrait exercer son pouvoir de décision indépendant, et elle serait donc en mesure de les forcer à accepter les mêmes contrats insensés et presque entièrement expurgés de Pfizer, Moderna et AstraZeneca, qui renoncent de toute façon à toute responsabilité et interdisent en outre aux pays de modifier leurs propres lois en ce qui concerne la responsabilité !
Les acheteurs doivent « indemniser, défendre et dégager de toute responsabilité Pfizer … contre toute poursuite, réclamation, action, demande, perte, dommage, responsabilité, règlement, pénalité, amende, coût et dépense … découlant de, en rapport avec, ou résultant du vaccin »
Principales objections notables et absence d’objections de la part des examinateurs de l’EMA.
La section relative à la chimie, à la fabrication et aux contrôles (CMC) de la demande de licence de produit biologique est le principal pilier de l’approbation réglementaire. Elle décrit le processus de fabrication et la conformité aux bonnes pratiques de fabrication (BPF), ainsi qu’à un vaste ensemble de lois et de règlements visant à garantir la pureté, l’efficacité, la cohérence et la sécurité des médicaments et des produits biologiques produits en masse. Les données relatives à la sécurité et à l’efficacité issues des essais cliniques sont inutiles si le fabricant ne peut garantir aux autorités de réglementation et à la communauté médicale que 1) le produit en question a été utilisé dans les essais cliniques conformément aux spécifications, 2) le produit est fabriqué de manière cohérente, pure, de haute qualité, reproductible, avec un processus de fabrication et des étapes de contrôle bien caractérisés et prévisibles, 3) le même produit que celui qui a été testé sera distribué dans le commerce.
Des problèmes ont été identifiés dans la section CMC de la demande de Pfizer :

Les évaluateurs CMC n’étaient pas satisfaits du calendrier donné pour l’évaluation, car il violait tous les délais normaux et tous les délais accélérés, par une grande marge. La solution consistait donc à « pousser » les évaluateurs. Cela n’a permis d’atteindre qu’un seul objectif : forcer les personnes qui auraient pu soulever des questions à s’épuiser pour qu’elles abandonnent et acceptent de travailler. Après tout, les dirigeants savaient très bien que l’examen réglementaire n’avait aucune signification et aucune implication sur la fausse « approbation », et qu’il aurait lieu quoi qu’il arrive. Au Royaume-Uni, la MHRA a déjà admis qu’elle ne disposait d’aucune délégation formelle d’autorité pour examiner et approuver ces injections, et je suis prêt à parier que l’EMA ne disposait pas non plus d’une telle autorité.
Les évaluateurs de bas niveau de la CMC ne le savaient pas et travaillaient très dur et très probablement de bonne foi. Fin novembre, ils ont soulevé plus de 140 objections formelles à l’encontre de la présentation CMC de Pfizer, qui comportait encore de nombreuses lacunes et informations manquantes. Pour mémoire, 10 à 15 objections réglementaires empêchent normalement une demande pharmaceutique d’aller de l’avant jusqu’à ce que les objections soient résolues. Trois objections majeures, c’est-à-dire des signaux d’alarme formels, sont discutées spécifiquement dans les courriels ci-dessous. J’ai, avec d’autres, beaucoup écrit sur l’objection majeure n° 2 (manque d’intégrité de l’ARNm). Voici un courriel de l’un des évaluateurs, Evdokia Korakianiti, et une réponse d’Alexis Nolte discutant du problème et de l’impact (totalement inconnu et potentiellement très troublant) sur l’efficacité et la sécurité du produit :

Le problème de la dégradation de l’ARNm a également été abordé par les spécialistes de la qualité CMC Ton van der Stappen, expert biopharmaceutique principal auprès du Conseil d’évaluation des médicaments (basé aux Pays-Bas) et spécialiste de la qualité pour l’EMA :

et Brian Dooley, un autre spécialiste de la qualité pharmaceutique à l’EMA :

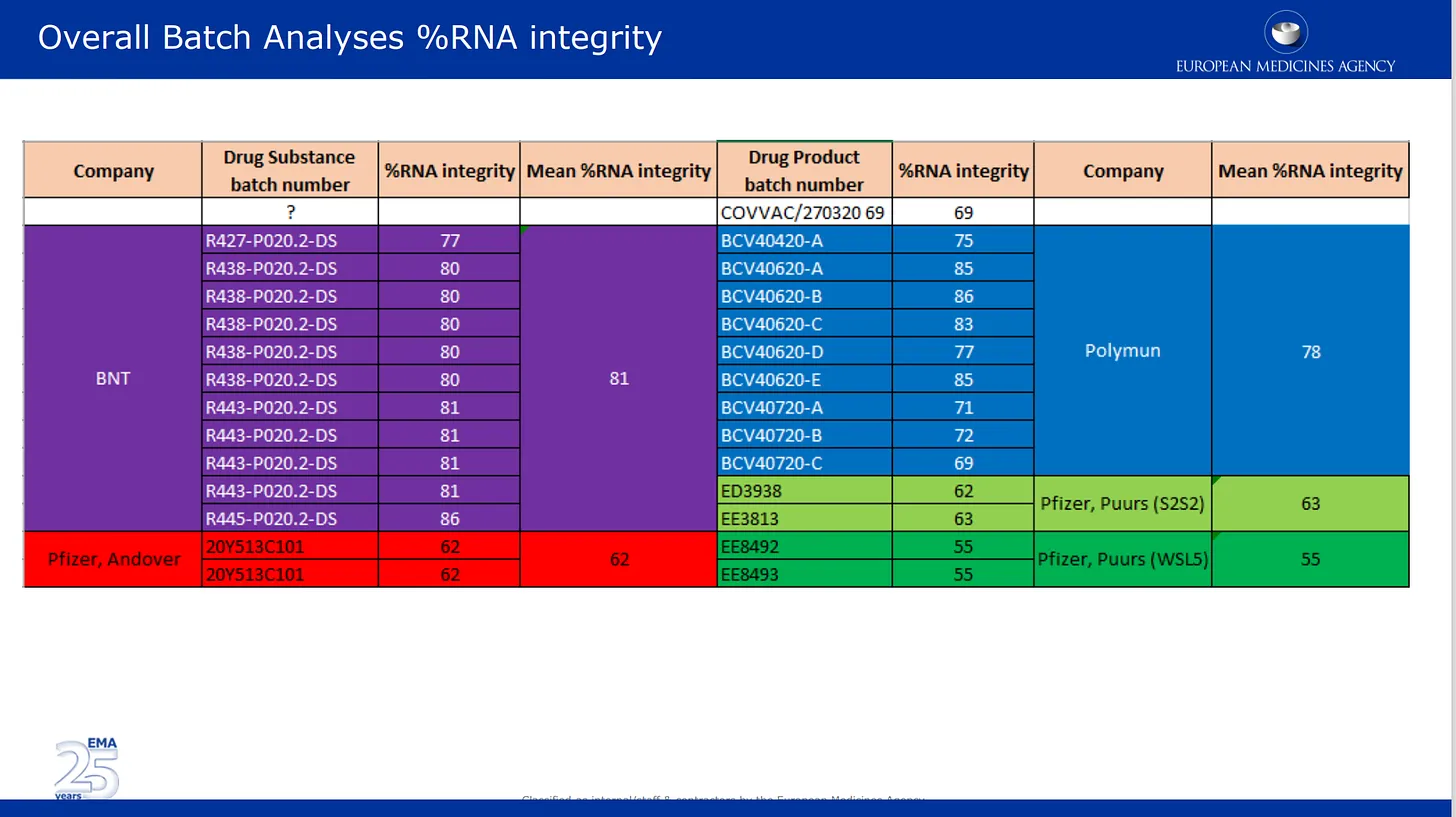
Les images ci-dessous sont tirées de l’examen de qualité qu’ils ont soumis à l’EMA. La première image traite du problème, désormais bien documenté, de la dégradation de l’ARNm dans différents lots du produit de Pfizer. Ici, les résultats d’analyse des lots fournis par Pfizer sont répertoriés et codés par couleur selon les différents sites de fabrication, ainsi que par substance médicamenteuse et catégorie de produit pharmaceutique. Substance médicamenteuse = composants actifs du produit (ARNm seul) et produit médicamenteux est la substance formulée dans les lipides et autres ingrédients. Le % d’intégrité de l’ARNm décrit le % d’ARNm « pleine longueur » détecté dans un lot. L’autre partie du lot était composée de fragments inconnus dont les propriétés ou l’impact sur la sécurité sont inconnus. Il convient de noter qu’aucune vérification indépendante n’a été effectuée par les autorités de réglementation, qui se sont contentées de reprendre les chiffres fournis par Pfizer/BioNTech.
Il semble que ces deux consultants scientifiques aient examiné et accepté les fausses images des résultats du Western blot soumis par Pfizer à l’EMA – les voici dans leur propre présentation PowerPoint du 24 novembre 2020. Lisez la note sous la diapositive – ils acceptent ces images comme étant réelles, même si ces deux réviseurs devraient être mieux informés. Pourquoi ne se sont-ils pas opposés à cela ? Voici ce que dit la note :
La taille de la protéine après l’expression in vitro de la substance médicamenteuse BNT162b2 a été déterminée par Western blot. La taille de la protéine exprimée a été confirmée comme étant comparable pour les trois lots du processus 1 et le lot du processus 2. La figure 3.2.S.2.6-15 montre que la taille de la protéine exprimée correspond à la taille attendue de la substance médicamenteuse BNT162b2 et est comparable pour tous les lots testés. En outre, les niveaux d’expression relative sont comparables pour tous les lots, comme le montre l’intensité comparable des bandes à chaque niveau de charge dans tous les lots.

Peut-être que des journalistes sérieux devraient contacter les docteurs van der Stappen et Dooley, ainsi que Mme Korakianiti et d’autres personnes mentionnées ici pour obtenir des commentaires.
Il ressort clairement des réponses données par Evdokia Korakianiti que la direction de l’EMA a renoncé aux armes et s’est appuyée sur des « données que seule la FDA a vues », mais qu’elle est « optimiste » et que la FDA a déclaré que la rupture de l’ARNm était une « préoccupation théorique ». Vraiment ? Y a-t-il des données à l’appui de cette affirmation ou non ? Voici les courriels indiquant que les objections majeures ont été formellement rédigées, puis ignorées par l’EMA puisque le produit a été expédié commercialement quelques semaines plus tard. Les conditions de l’AMC n’ont jamais été remplies.
Cela confirme ce que nous savions déjà : ni l’EMA (ni la FDA, ni Santé Canada, ni la MHRA, ni aucun autre organisme de réglementation) n’avait d’autorité réelle sur ces produits, ni d’impact sur leur déploiement auprès d’un public peu méfiant. Tout cela n’était que du théâtre, du début à la fin.
Voici les trois principales objections qui n’ont toujours pas été levées à ce jour :

Et voici quelques signes de la main et l’acceptation des affirmations de la FDA sans aucune remise en question ou évaluation formelle des données par les régulateurs de l’EMA :

Ce que je peux dire pour conclure, c’est que j’ai dénombré environ 70 personnes différentes mentionnées dans le document et les courriels ayant fait l’objet d’une fuite, qui ont facilité cette tragique mascarade, à savoir l' »approbation » du produit le plus mortel jamais lâché sur le plus grand nombre de personnes, entraînant un nombre sans précédent de morts et de blessés dans le monde entier. Peut-être, à quelques exceptions près, la plupart d’entre eux ont-ils été trompés en 2020 et n’ont pas compris qu’ils participaient à un crime de guerre et qu’ils signaient une fraude mortelle. Je pense que la plupart d’entre eux le savent maintenant, j’espère qu’ils sont suffisamment horrifiés par ce qu’ils ont rendu possible, et j’espère que ces personnes vont se manifester en tant que dénonciateurs et commencer à parler. Nous avons besoin de réponses.
Publié à l’origine par Due Diligence and Art
Suggérer une correction